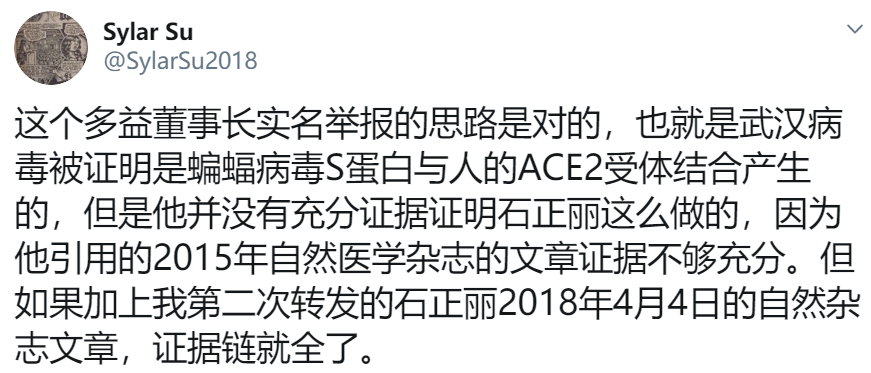
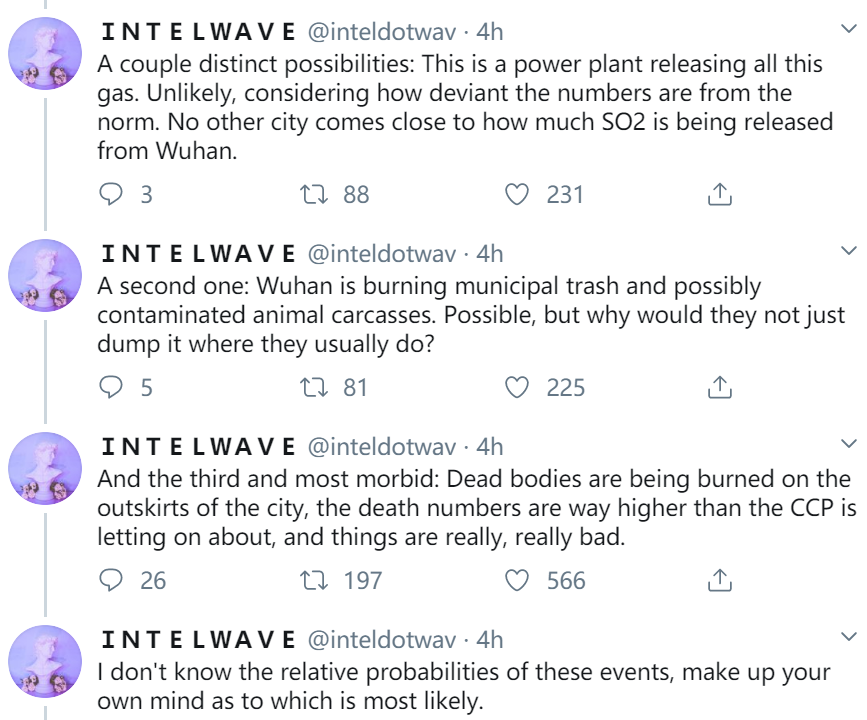
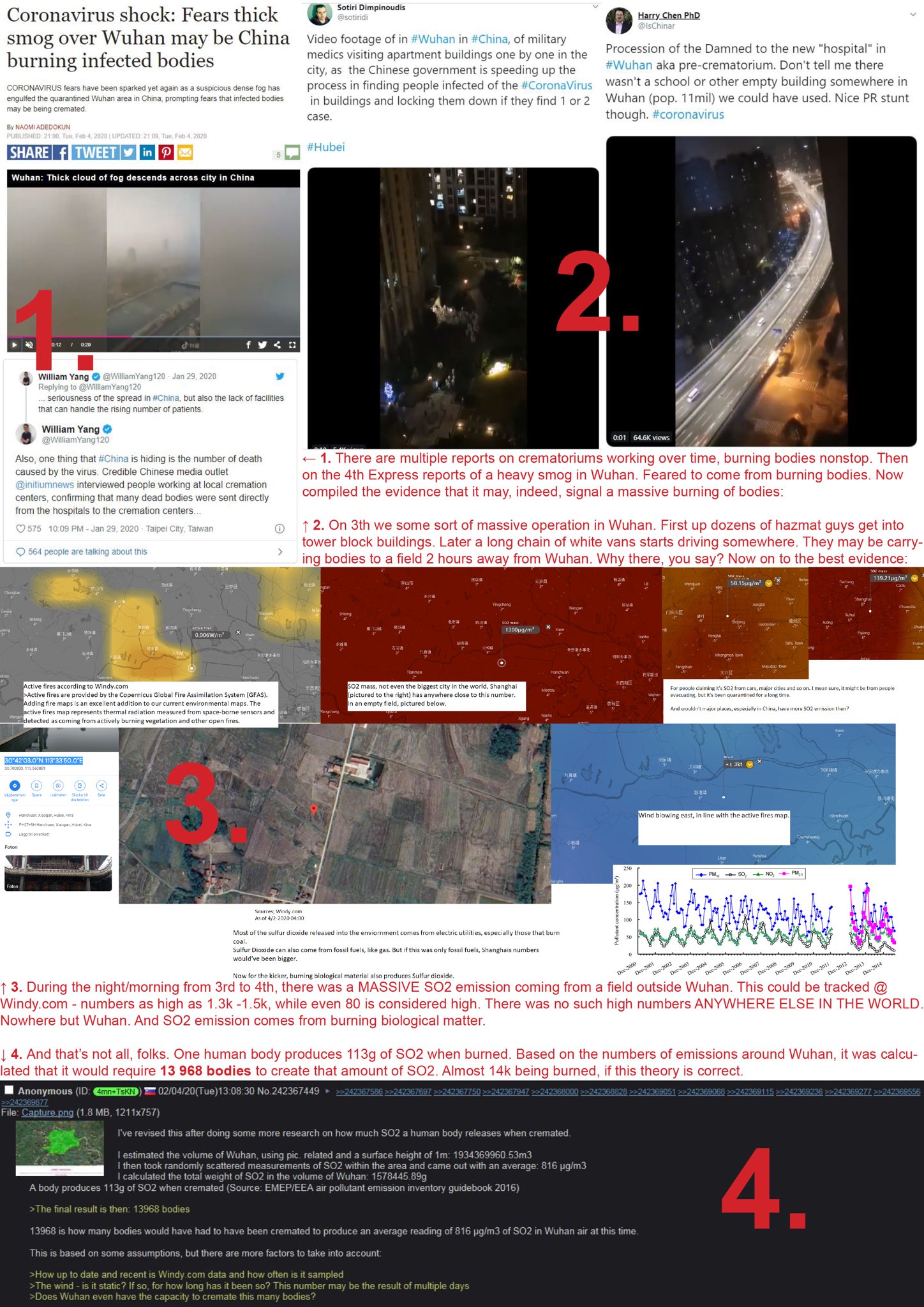
(Natural News) Every virology lab in the world that has run a genomic analysis of the coronavirus now knows that the coronavirus was engineered by human scientists. The proof is in the virus itself: The tools for genetic insertion are still present as remnants in the genetic code. Since these unique gene sequences don’t occur by random chance, they’re proof that this virus was engineered by scientists in a lab.
But the WHO and CDC are covering up this inconvenient fact in order to protect communist China and its biological weapons program, since no government wants the public to know the full truth about how frequently government-run labs experience outbreaks. Decades ago, for example, the U.S. Army ran an Ebola bioweapons lab in the United States, where a monkey infected one of the scientists there. The strain turned out to be infectious only in monkeys, not humans, so the world dodged a bullet, but the U.S. Army “nuked” the entire facility with chemical bombs, killing all the monkeys and wiping out any last remnant of the virus on U.S. soil.
You can read the full details of that incident in the book The Hot Zone by Richard Preston. We’ve also covered it at NaturalNews.com, where this book description is reprinted:
In 1989, Reston, VA — one of the most famous U.S. planned communities located about 10 miles from Washington DC — stood at the epicenter of a potential biological disaster. This well-known story was narrated by Richard Preston in a bone chilling account related to the recognition and containment of a devastating tropical filovirus at a monkey facility — the Reston Primate Quarantine Unit.
That outbreak occurred because Ebola was found to be spreading through the air ducts, confirming that Ebola can spread through the air. This simple fact was vigorously covered up by the entire medical establishment during the Ebola scare in the United States many years later, where the CDC transported an infected patient to a hospital in Dallas, subsequently infecting a nurse who was treated with highly toxic chemicals that caused permanent kidney damage (she later sued the hospital for the damage she suffered).
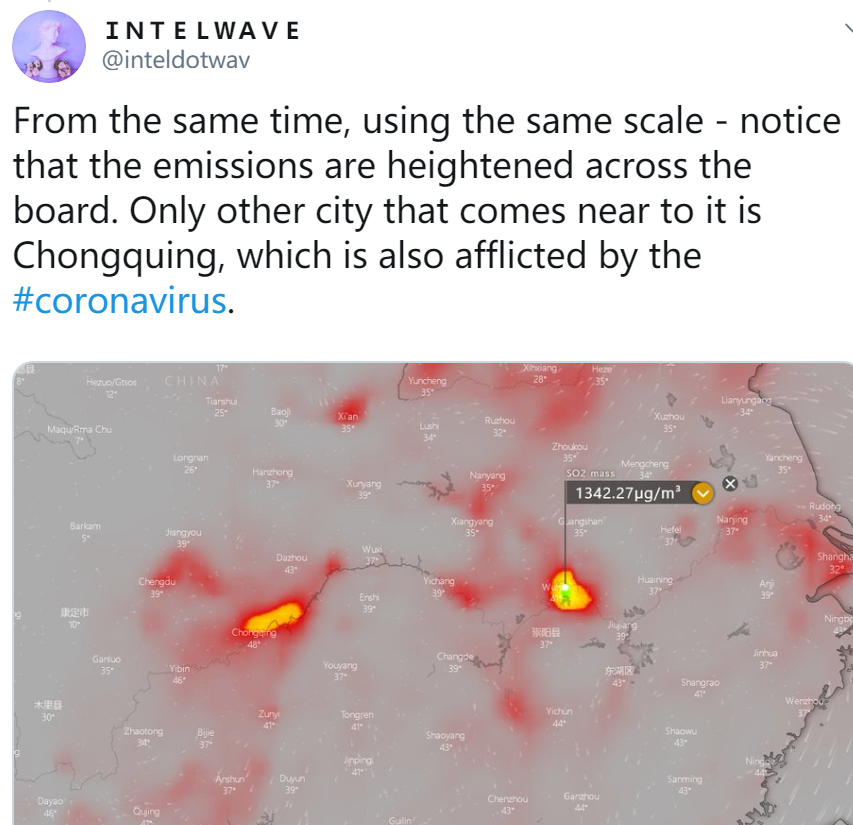
The reason this is relevant is because in order to understand the coronavirus situation in China, we must first realize that virology research labs routinely experience lapses in containment. Even the United States has failed to contain deadly viral strains when trying to study them. China’s BSL-4 labs have experienced multiple accidental releases of SARS strains, and this new coronavirus is now confirmed to be an engineered strain that was either used in bioweapons research or vaccine experiments.
The genomic coding in the virus is not natural, in other words. Just as you would never encounter a snake in the desert that’s writing a book containing words and grammatical structure, the genetic sequences now identified in the coronavirus strain are, without question, proof that human engineers have been tinkering with the strain.
How to genetically engineer viruses: the pShuttle vector
One of the tools used to accomplish this genetic engineering is called pShuttle. It’s a genetic tool set that can carry a payload of genes to be inserted into the target virus.
Researchers engaged in genetic engineering can purchase the pShuttle sequence from online retailers such as AddGenes.org, which sells the sequence for $75, shipped in “bacteria as agar stab.”
The following map outlines the complete gene sequence of the pShuttle tool:
The summary of the paper describes, “a strategy that simplifies the generation and production of such viruses.” Here’s how the process works to achieve genetic engineering of viruses:
A recombinant adenoviral plasmid is generated with a minimum of enzymatic manipulations, using homologous recombination in bacteria rather than in eukaryotic cells. After transfections of such plasmids into a mammalian packaging cell line, viral production is conveniently followed with the aid of green fluorescent protein, encoded by a gene incorporated into the viral backbone. Homogeneous viruses can be obtained from this procedure without plaque purification.
The paper describes how this approach will, “expedite the process of generating and testing recombinant adenoviruses.”
During this process, of course, the pShuttle leaves behind unique code, a “fingerprint” of the genetic modification. It is this fingerprint that has now been identified in the coronavirus.
As revealed by genomics researcher James Lyons-Weiler in this bombshell analysis article, the pShuttle genetic code is found in the coronavirus that’s circulating in the wild.
This is proof that the virus has been engineered by human scientists.
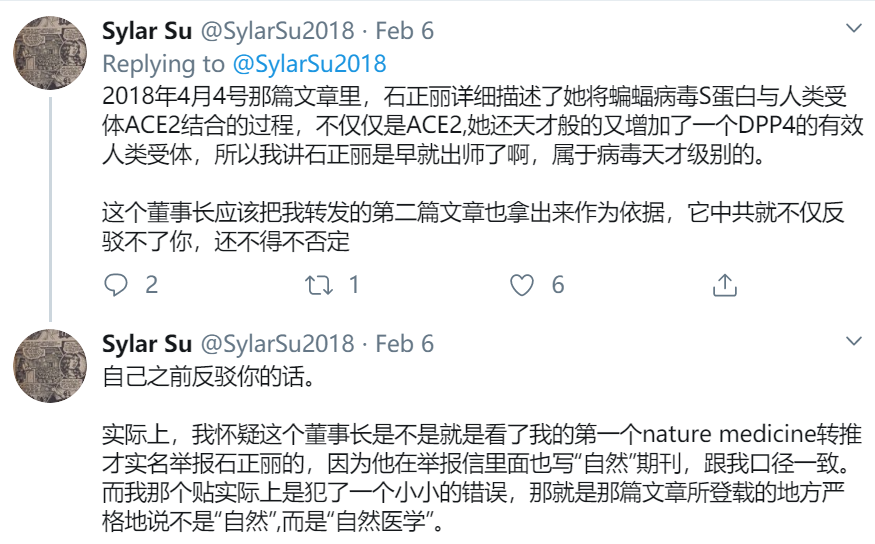
“IPAK researchers found a sequence similarity between a pShuttle-SN recombination vector sequence and INS1378,” writes Lyons-Weiler for IPAK:
Another gene sequence also shows a 92% match with the Spike protein from the SARS coronavirus:
The pShuttle vector was used to insert SARS genes into the coronavirus, a process that makes it deadly to humans. “The very researchers conducting studies on SARS vaccines have cautioned repeatedly against human trials,” warns Lyons-Weiler:
The disease progression in of 2019-nCoV is consistent with those seen in animals and humans vaccinated against SARS and then challenged with re-infection. Thus, the hypothesis that 2019-nCoV is an experimental vaccine type must be seriously considered.
He also warns about, “studies that have reported serious immunopathology in animals – rats, ferrets, and monkeys – in which animals vaccinated against coronoviruses tended to have extremely high rates of respiratory failure upon subsequent exposure in the study when challenged with the wild-type coronavirus.”
He concludes:
If the Chinese government has been conducting human trials against SARS. MERS, or other coronviruses using recombined viruses, they may have made their citizens far more susceptible to acute respiratory distress syndrome upon infection with 2019-nCoV coronavirus.
Another doctor from Beijing Medical University warns the virus appears to be genetically engineered
Lyons-Weiler is not alone in his assessment of the genetic engineering origins of the coronavirus. Dr. Yuhong Dong, who holds a doctorate degree in infectious diseases from Beijing University, writes in The Epoch Times:
Based on recently published scientific papers, this new coronavirus has unprecedented virologic features that suggest genetic engineering may have been involved in its creation. The virus presents with severe clinical features, thus it poses a huge threat to humans. It is imperative for scientists, physicians, and people all over the world, including governments and public health authorities, to make every effort to investigate this mysterious and suspicious virus in order to elucidate its origin and to protect the ultimate future of the human race.
Dr. Yuhong reminds us that a Jan. 30 science paper published in The Lancet concludes that, “recombination is probably not the reason for emergence of this virus.” In other words, this did not occur through natural mutations in the wild.
He also points to a Jan. 27th study by five Greek scientists who also concluded the coronavirus has no lineage to other viruses in the “family tree” that’s found in the wild. He writes:
A Jan. 27 2020, study by 5 Greek scientists analyzed the genetic relationships of 2019-nCoV and found that “the new coronavirus provides a new lineage for almost half of its genome, with no close genetic relationships to other viruses within the subgenus of sarbecovirus,” and has an unusual middle segment never seen before in any coronavirus. All this indicates that 2019-nCoV is a brand-new type of coronavirus. The study’s authors rejected the original hypothesis that 2019-nCoV originated from random natural mutations between different coronaviruses.
“ No bats were sold or found at the
Huanan seafood market ”
Dr. Yuhong writes about The Lancet study by authors Roujian Lu et al., from the China Key Laboratory of Biosafety, National Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, repeating a quote from that paper:
First, the outbreak was first reported in late December 2019, when most bat species in Wuhan are hibernating. Second, no bats were sold or found at the Huanan seafood market, whereas various non-aquatic animals (including mammals) were available for purchase. Third, the sequence identity between 2019-nCoV and its close relatives bat-SL-CoVZC45 and bat-SL-CoVZXC21 was less than 90%. Hence, bat-SL-CoVZC45 and bat-SL-CoVZXC21 are not direct ancestors of 2019-nCoV.
In other words, it isn’t from bats.
That means the entire mainstream media is lying to us about the real origins of the coronavirus.
That same paper goes on to underscore the misinformation in the official explanation, stating, “Many of the initially confirmed 2019-nCoV cases—27 of the first 41 in one report, 26 of 47 in another—were connected to the Wuhan market, but up to 45%, including the earliest handful, were not. This raises the possibility that the initial jump into people happened elsewhere.”
Both Lu (in The Lancet paper linked above) and Lyons-Weiler point to the presence of a SARS binding protein sequence in the coronavirus that allows it to easily infect human cells. As explained in The Epoch Times:
…despite considerable genetics distance between the Wuhan CoV and the human-infecting SARS-CoV, and the overall low homology of the Wuhan CoV S-protein to that of SARS-CoV, the Wuhan CoV S-protein had several patches of sequences in the receptor binding (RBD) domain with a high homology to that of SARS-CoV. The residues at positions 442, 472, 479, 487, and 491 in SARS-CoV S-protein were reported to be at receptor complex interface and considered critical for cross species and human-to-human transmission of SARS-CoV. So to our surprise, despite replacing four out of five important interface amino acid residues, the Wuhan CoV S-protein was found to have a significant binding affinity to human ACE2. …The Wuhan CoV S-protein and SARS-CoV S-protein shared an almost identical 3-D structure in the RBD domain, thus maintaining similar van der Waals and electrostatic properties in the interaction interface. Thus the Wuhan CoV is still able to pose a significant public health risk for human transmission via the S protein–ACE2 binding pathway. (emphasis added)
As Dr. Yuhong asks, “How could this novel virus be so intelligent as to mutate precisely at selected sites while preserving its binding affinity to the human ACE2 receptor? How did the virus change just four amino acids of the S-protein? Did the virus know how to use Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) to make sure this would happen?”
It couldn’t happen by chance, in other words. The coronavirus is not a random mutation in the wild. It was engineered.
Many other scientists around the world are now investigating the gene sequences found in the coronavirus, and they are increasingly concluding that elements of the virus have been engineered.
Many of those scientists are being threatened and censored. One paper has so far been forced to be withdrawn and revised, no doubt to remove the key conclusions that point to the genetic engineering origins of the coronavirus, but the proof of its engineering cannot be denied forever.
Either the coronavirus was genetically engineered, or the science establishment is going to have to throw out the entire field of genomics research and claim it isn’t real
Eventually, the science establishment is either going to have to conclude that this coronavirus strain was engineered, or that all the laws of genetics science don’t work, and gene sequencing is imaginary (sort of like transgenderism by the “progressive” Left, which has already abandoned biological reality).
So far they’ve tried to bamboozle the public into believing this is all some sort of accident from Mother Nature, but that has only worked because most of the public doesn’t understand enough science to counter the official propaganda. However, there are more than enough independent scientists around the world to prove that this pandemic strain was engineered by humans. More evidence is coming out each day.
Interestingly, as this article is going to press, all the official numbers of infections and deaths from coronavirus have been frozen for about 14 hours and counting, almost as if every nation of the world has agreed to stop reporting new numbers. This may be a temporary situation that gets resolved in the next few hours, but it’s highly suspicious. For the last week, we’ve been getting new updates about every 12 hours or sooner, and we’ve never seen the count frozen for this long.
At the same time, an 11th case of coronavirus has now been confirmed by the CDC in the United States, revealing that infections are continuing to spread in the USA, despite the efforts of the CDC to contain the outbreak.
Lab-Made Coronavirus Triggers DebateThe creation of a chimeric SARS-like virus has scientists discussing the risks of gain-of-function research.Nov 16, 2015JEF AKST
MERS coronavirusFLICKR, NIAID
Ralph Baric, an infectious-disease researcher at the University of North Carolina at Chapel Hill, last week (November 9) published a study on his team’s efforts to engineer a virus with the surface protein of the SHC014 coronavirus, found in horseshoe bats in China, and the backbone of one that causes human-like severe acute respiratory syndrome (SARS) in mice. The hybrid virus could infect human airway cells and caused disease in mice, according to the team’s results, which were published in Nature Medicine.
The results demonstrate the ability of the SHC014 surface protein to bind and infect human cells, validating concerns that this virus—or other coronaviruses found in bat species—may be capable of making the leap to people without first evolving in an intermediate host, Nature reported. They also reignite a debate about whether that information justifies the risk of such work, known as gain-of-function research. “If the [new] virus escaped, nobody could predict the trajectory,” Simon Wain-Hobson, a virologist at the Pasteur Institute in Paris, told Nature.
In October 2013, the US government put a stop to all federal funding for gain-of-function studies, with particular concern rising about influenza, SARS, and Middle East respiratory syndrome (MERS). “NIH [National Institutes of Health] has funded such studies because they help define the fundamental nature of human-pathogen interactions, enable the assessment of the pandemic potential of emerging infectious agents, and inform public health and preparedness efforts,” NIH Director Francis Collins said in a statement at the time. “These studies, however, also entail biosafety and biosecurity risks, which need to be understood better.”
Baric’s study on the SHC014-chimeric coronavirus began before the moratorium was announced, and the NIH allowed it to proceed during a review process, which eventually led to the conclusion that the work did not fall under the new restrictions, Baric told Nature. But some researchers, like Wain-Hobson, disagree with that decision.
The debate comes down to how informative the results are. “The only impact of this work is the creation, in a lab, of a new, non-natural risk,” Richard Ebright, a molecular biologist and biodefence expert at Rutgers University, told Nature.
But Baric and others argued the study’s importance. “[The results] move this virus from a candidate emerging pathogen to a clear and present danger,” Peter Daszak, president of the EcoHealth Alliance, which samples viruses from animals and people in emerging-diseases hotspots across the globe, told Nature.
给你看看美国的科学(Science)杂志发出来的东西看看,“The role of Huanan Seafood Wholesale Market in Wuhan, China, in spreading 2019-nCoV remains murky, though such sequencing, combined with sampling the market’s environment for the presence of the virus, is clarifying that it indeed had an important early role in amplifying the outbreak. The viral sequences, most researchers say, also knock down the idea the pathogen came from a virology institute in Wuhan.” 见于文章题目“
Mining coronavirus genomes for clues to the outbreak’s origins”----文章链接:https://www.sciencemag.org/news/2020/01/mining-coronavirus-genomes-clues-outbreak-s-origins
The discovery of SARS-like coronavirus in bats suggests that bats could be the natural reservoir of SARS-CoV. However, previous studies indicated the angiotensin-converting enzyme 2 (ACE2) protein, a known SARS-CoV receptor, from a horseshoe bat was unable to act as a functional receptor for SARS-CoV. Here, we extended our previous study to ACE2 molecules from seven additional bat species and tested their interactions with human SARS-CoV spike protein using both HIV-based pseudotype and live SARS-CoV infection assays. The results show that ACE2s of Myotis daubentoni and Rhinolophus sinicus support viral entry mediated by the SARS-CoV S protein, albeit with different efficiency in comparison to that of the human ACE2. Further, the alteration of several key residues either decreased or enhanced bat ACE2 receptor efficiency, as predicted from a structural modeling study of the different bat ACE2 molecules. These data suggest that M. daubentoni and R. sinicus are likely to be susceptible to SARS-CoV and may be candidates as the natural host of the SARS-CoV progenitor viruses. Furthermore, our current study also demonstrates that the genetic diversity of ACE2 among bats is greater than that observed among known SARS-CoV susceptible mammals, highlighting the possibility that there are many more uncharacterized bat species that can act as a reservoir of SARS-CoV or its progenitor viruses. This calls for continuation and expansion of field surveillance studies among different bat populations to eventually identify the true natural reservoir of SARS-CoV.
Introduction
Severe acute respiratory syndrome coronavirus (SARS-CoV) is the aetiological agent responsible for the SARS outbreaks during 2002–2003, which had a huge global impact on public health, travel and the world economy [4, 11]. The host range of SARS-CoV is largely determined by the specific and high-affinity interactions between a defined receptor-binding domain (RBD) on the SARS-CoV spike protein and its host receptor, angiontensin-converting enzyme 2 (ACE2) [6, 7, 9]. It has been hypothesized that SARS-CoV was harbored in its natural reservoir, bats, and was transmitted directly or indirectly from bats to palm civets and then to humans [10]. However, although the genetically related SARS-like coronavirus (SL-CoV) has been identified in horseshoe bats of the genus Rhinolophus [5, 8, 12, 18], its spike protein was not able to use the human ACE2 (hACE2) protein as a receptor [13]. Close examination of the crystal structure of human SARS-CoV RBD complexed with hACE2 suggests that truncations in the receptor-binding motif (RBM) region of SL-CoV spike protein abolish its hACE2-binding ability [7, 10], and hence the SL-CoV found recently in horseshoe bats is unlikely to be the direct ancestor of human SARS-CoV. Also, it has been shown that the human SARS-CoV spike protein and its closely related civet SARS-CoV spike protein were not able to use a horseshoe bat (R. pearsoni) ACE2 as a receptor [13], highlighting a critical missing link in the bat-to-civet/human transmission chain of SARS-CoV.
There are at least three plausible scenarios to explain the origin of SARS-CoV. First, some unknown intermediate hosts were responsible for the adaptation and transmission of SARS-CoV from bats to civets or humans. This is the most popular theory of SARS-CoV transmission at the present time [10]. Second, there is an SL-CoV with a very close relationship to the outbreak SARS-CoV strains in a non-bat animal host that is capable of direct transmission from reservoir host to human or civet. Third, ACE2 from yet to be identified bat species may function as an efficient receptor, and these bats could be the direct reservoir of human or civet SARS-CoV. Unraveling which scenario is most likely to have occurred during the 2002–2003 SARS epidemic is critical for our understanding of the dynamics of the outbreak and will play a key role in helping us to prevent future outbreaks. To this end, we have extended our studies to include ACE2 molecules from different bat species and examined their interaction with the human SARS-CoV spike protein. Our results show that there is great genetic diversity among bat ACE2 molecules, especially at the key residues known to be important for interacting with the viral spike protein, and that ACE2s of Myotis daubentoni and Rhinolophus sinicus from Hubei province can support viral entry.
LetterPublished: 04 April 2018Fatal swine acute diarrhoea syndrome caused by an HKU2-related coronavirus of bat originPeng Zhou, Hang Fan, […]Jing-Yun Ma
Nature volume 556, pages255–258(2018)Cite this article
26k Accesses
84 Citations
902 Altmetric
Metricsdetails
This article has been updated
Abstract
Cross-species transmission of viruses from wildlife animal reservoirs poses a marked threat to human and animal health1. Bats have been recognized as one of the most important reservoirs for emerging viruses and the transmission of a coronavirus that originated in bats to humans via intermediate hosts was responsible for the high-impact emerging zoonosis, severe acute respiratory syndrome (SARS)2,3,4,5,6,7,8,9,10. Here we provide virological, epidemiological, evolutionary and experimental evidence that a novel HKU2-related bat coronavirus, swine acute diarrhoea syndrome coronavirus (SADS-CoV), is the aetiological agent that was responsible for a large-scale outbreak of fatal disease in pigs in China that has caused the death of 24,693 piglets across four farms. Notably, the outbreak began in Guangdong province in the vicinity of the origin of the SARS pandemic. Furthermore, we identified SADS-related CoVs with 96–98% sequence identity in 9.8% (58 out of 591) of anal swabs collected from bats in Guangdong province during 2013–2016, predominantly in horseshoe bats (Rhinolophus spp.) that are known reservoirs of SARS-related CoVs. We found that there were striking similarities between the SADS and SARS outbreaks in geographical, temporal, ecological and aetiological settings. This study highlights the importance of identifying coronavirus diversity and distribution in bats to mitigate future outbreaks that could threaten livestock, public health and economic growth.
Main
The emergence of SARS in southern China in 2002, which was caused by a previously unknown coronavirus (SARS-CoV)11,12,13,14,15 and has led to more than 8,000 human infections and 774 deaths (http://www.who.int/csr/sars/en/), highlights two new frontiers in emerging infectious diseases. First, it demonstrates that coronaviruses are capable of causing fatal diseases in humans. Second, the identification of bats as the reservoir for SARS-related coronaviruses, and the fact that SARS-CoV3,4,5,6,7,8,9,10 probably originated in bats, firmly establishes that bats are an important source of highly lethal zoonotic viruses, such as Hendra, Nipah, Ebola and Marburg viruses16.
Here we report on a series of fatal swine disease outbreaks in Guangdong province, China, approximately 100 km from the location of the purported index case of SARS. Most strikingly, we found that the causative agent of this swine acute diarrhoea syndrome (SADS) is a novel HKU2-related coronavirus that is 98.48% identical in genome sequence to a bat coronavirus, which we detected in 2016 in bats in a cave in the vicinity of the index pig farm. This new virus (SADS-CoV) originated from the same genus of horseshoe bats (Rhinolophus) as SARS-CoV.
From 28 October 2016 onwards, a fatal swine disease outbreak was observed in a pig farm in Qingyuan, Guangdong province, China, very close to the location of the first known index case of SARS in 2002, who lived in Foshan (Extended Data Fig. 1a). Porcine epidemic diarrhoea virus (PEDV, a coronavirus) had caused prior outbreaks at this farm, and was detected in the intestines of deceased piglets at the start of the outbreak. However, PEDV could no longer be detected in deceased piglets after 12 January 2017, despite accelerating mortality (Fig. 1a), and extensive testing for other common swine viruses yielded no results (Extended Data Table 1). These findings suggested that this was an outbreak of a novel disease. Clinical signs are similar to those caused by other known swine enteric coronaviruses17, 18 and include severe and acute diarrhoea and acute vomiting, leading to death due to rapid weight loss in newborn piglets that are less than five days of age. Infected piglets died 2–6 days after disease onset, whereas infected sows suffered only mild diarrhoea and most sows recovered within two days. The disease caused no signs of febrile illness in piglets or sows. The mortality rate was as high as 90% in piglets that were five days or younger, whereas in piglets that were older than eight days, the mortality dropped to 5%. Subsequently, SADS-related outbreaks were found in three additional pig farms within 20–150 km of the index farm (Extended Data Fig. 1a) and, by 2 May 2017, the disease had caused the death of 24,693 piglets at these four farms (Fig. 1a). In farm A alone, 64% (4,659 out of 7,268) of all piglets that were born in February died. The outbreak has abated, and measures that were taken to control SADS included separation of sick sows and piglets from the rest of the herd. A qPCR test described below was used as the main diagnostic tool to confirm SADS-CoV infection.














![addgene-pshuttle-gene-sequence Irrefutable: The coronavirus was engineered by scientists in a lab using well documented genetic engineering vectors that leave behind a “fingerprint” [your]NEWS](https://s3-ap-northeast-1.amazonaws.com/news.guo.offload.media/wp-content/uploads/2020/02/08024353/1-10.png)
![addgene-pshuttle-gene-sequence Irrefutable: The coronavirus was engineered by scientists in a lab using well documented genetic engineering vectors that leave behind a “fingerprint” [your]NEWS](https://s3-ap-northeast-1.amazonaws.com/news.guo.offload.media/wp-content/uploads/2020/02/08024435/2-9.png)
![addgene-pshuttle-gene-sequence Irrefutable: The coronavirus was engineered by scientists in a lab using well documented genetic engineering vectors that leave behind a “fingerprint” [your]NEWS](https://s3-ap-northeast-1.amazonaws.com/news.guo.offload.media/wp-content/uploads/2020/02/08024452/3-3.png)
![addgene-pshuttle-gene-sequence Irrefutable: The coronavirus was engineered by scientists in a lab using well documented genetic engineering vectors that leave behind a “fingerprint” [your]NEWS](https://s3-ap-northeast-1.amazonaws.com/news.guo.offload.media/wp-content/uploads/2020/02/08024506/4-5.png)
![addgene-pshuttle-gene-sequence Irrefutable: The coronavirus was engineered by scientists in a lab using well documented genetic engineering vectors that leave behind a “fingerprint” [your]NEWS](https://s3-ap-northeast-1.amazonaws.com/news.guo.offload.media/wp-content/uploads/2020/02/08024515/5-7.png)
![addgene-pshuttle-gene-sequence Irrefutable: The coronavirus was engineered by scientists in a lab using well documented genetic engineering vectors that leave behind a “fingerprint” [your]NEWS](https://s3-ap-northeast-1.amazonaws.com/news.guo.offload.media/wp-content/uploads/2020/02/08024524/6-3.png)
![addgene-pshuttle-gene-sequence Irrefutable: The coronavirus was engineered by scientists in a lab using well documented genetic engineering vectors that leave behind a “fingerprint” [your]NEWS](https://s3-ap-northeast-1.amazonaws.com/news.guo.offload.media/wp-content/uploads/2020/02/08024549/7-4.png)


![addgene-pshuttle-gene-sequence Irrefutable: The coronavirus was engineered by scientists in a lab using well documented genetic engineering vectors that leave behind a “fingerprint” [your]NEWS](https://www.naturalnews.com/wp-content/uploads/2020/02/addgene-pshuttle-gene-sequence.png "addgene-pshuttle-gene-sequence Irrefutable: The coronavirus was engineered by scientists in a lab using well documented genetic engineering vectors that leave behind a “fingerprint” [your]NEWS")
![addgene-pshuttle-gene-sequence Irrefutable: The coronavirus was engineered by scientists in a lab using well documented genetic engineering vectors that leave behind a “fingerprint” [your]NEWS](https://www.naturalnews.com/wp-content/uploads/2020/02/pshuttle-gene-map.png "addgene-pshuttle-gene-sequence Irrefutable: The coronavirus was engineered by scientists in a lab using well documented genetic engineering vectors that leave behind a “fingerprint” [your]NEWS")
![addgene-pshuttle-gene-sequence Irrefutable: The coronavirus was engineered by scientists in a lab using well documented genetic engineering vectors that leave behind a “fingerprint” [your]NEWS](https://www.naturalnews.com/images/Recombinant-Adenoviruses-1.jpg "addgene-pshuttle-gene-sequence Irrefutable: The coronavirus was engineered by scientists in a lab using well documented genetic engineering vectors that leave behind a “fingerprint” [your]NEWS")
![addgene-pshuttle-gene-sequence Irrefutable: The coronavirus was engineered by scientists in a lab using well documented genetic engineering vectors that leave behind a “fingerprint” [your]NEWS](https://www.naturalnews.com/images/Recombinant-Adenoviruses-4.jpg "addgene-pshuttle-gene-sequence Irrefutable: The coronavirus was engineered by scientists in a lab using well documented genetic engineering vectors that leave behind a “fingerprint” [your]NEWS")
![addgene-pshuttle-gene-sequence Irrefutable: The coronavirus was engineered by scientists in a lab using well documented genetic engineering vectors that leave behind a “fingerprint” [your]NEWS](https://www.naturalnews.com/wp-content/uploads/2020/02/pShuttle-SN-sequence-plot.png "addgene-pshuttle-gene-sequence Irrefutable: The coronavirus was engineered by scientists in a lab using well documented genetic engineering vectors that leave behind a “fingerprint” [your]NEWS")
![addgene-pshuttle-gene-sequence Irrefutable: The coronavirus was engineered by scientists in a lab using well documented genetic engineering vectors that leave behind a “fingerprint” [your]NEWS](https://www.naturalnews.com/wp-content/uploads/2020/02/sars-spike-protein-match.png "addgene-pshuttle-gene-sequence Irrefutable: The coronavirus was engineered by scientists in a lab using well documented genetic engineering vectors that leave behind a “fingerprint” [your]NEWS")
![addgene-pshuttle-gene-sequence Irrefutable: The coronavirus was engineered by scientists in a lab using well documented genetic engineering vectors that leave behind a “fingerprint” [your]NEWS](https://www.naturalnews.com/images/BioRxiv-Full-Genome-Coronavirus.jpg "addgene-pshuttle-gene-sequence Irrefutable: The coronavirus was engineered by scientists in a lab using well documented genetic engineering vectors that leave behind a “fingerprint” [your]NEWS")































 MERS coronavirusFLICKR, NIAID
MERS coronavirusFLICKR, NIAID