於是,Vineet D. Menachery教授等利用SARS-CoV的反向遺傳系統,從中提取並鑑定了一種嵌合病毒,該病毒在適應小白鼠的SARS-CoV主幹中可表達出蝙蝠冠狀病毒SHC014的刺突。結果表明,該新型冠狀病毒能利用SARS的人類細胞受體——血管緊張素轉換酶II(ACE2)的多個同源基因,在人類呼吸道原代細胞中有效複製,並在體外獲得與SARS傳染性同等的效果。
如前所述,野生型和嵌合病毒均來自 SARS-CoV Urbani 或相應的小鼠適應性(SARS-CoV MA15)感染性克隆(ic)病毒細胞。通過提取含有 SHC014 刺突序列的質粒,並連接到 MA15 感染性克隆的 E 和 F 質粒中。設計該克隆並從 Bio Basic 購買六種連續 cDNA,並使用側接獨特的 II 類限制性核酸內切酶位點(BglI)的公開序列。
Ralph Baric, an infectious-disease researcher at the University of North Carolina at Chapel Hill, last week (November 9) published a study on his team’s efforts to engineer a virus with the surface protein of the SHC014 coronavirus, found in horseshoe bats in China, and the backbone of one that causes human-like severe acute respiratory syndrome (SARS) in mice. The hybrid virus could infect human airway cells and caused disease in mice, according to the team’s results, which were published in Nature Medicine.
The results demonstrate the ability of the SHC014 surface protein to bind and infect human cells, validating concerns that this virus—or other coronaviruses found in bat species—may be capable of making the leap to people without first evolving in an intermediate host, Nature reported. They also reignite a debate about whether that information justifies the risk of such work, known as gain-of-function research. “If the [new] virus escaped, nobody could predict the trajectory,” Simon Wain-Hobson, a virologist at the Pasteur Institute in Paris, told Nature.
In October 2013, the US government put a stop to all federal funding for gain-of-function studies, with particular concern rising about influenza, SARS, and Middle East respiratory syndrome (MERS). “NIH [National Institutes of Health] has funded such studies because they help define the fundamental nature of human-pathogen interactions, enable the assessment of the pandemic potential of emerging infectious agents, and inform public health and preparedness efforts,” NIH Director Francis Collins said in a statement at the time. “These studies, however, also entail biosafety and biosecurity risks, which need to be understood better.”
Baric’s study on the SHC014-chimeric coronavirus began before the moratorium was announced, and the NIH allowed it to proceed during a review process, which eventually led to the conclusion that the work did not fall under the new restrictions, Baric told Nature. But some researchers, like Wain-Hobson, disagree with that decision.
The debate comes down to how informative the results are. “The only impact of this work is the creation, in a lab, of a new, non-natural risk,” Richard Ebright, a molecular biologist and biodefence expert at Rutgers University, told Nature.
But Baric and others argued the study’s importance. “[The results] move this virus from a candidate emerging pathogen to a clear and present danger,” Peter Daszak, president of the EcoHealth Alliance, which samples viruses from animals and people in emerging-diseases hotspots across the globe, told Nature.
*******************************
bats are an important source of highly lethal zoonotic viruses, such as Hendra, Nipah, Ebola and Marburg viruses16.
Here we report on a series of fatal swine disease outbreaks in Guangdong province, China, approximately 100 km from the location of the purported index case of SARS. Most strikingly, we found that the causative agent of this swine acute diarrhoea syndrome (SADS) is a novel HKU2-related coronavirus that is 98.48% identical in genome sequence to a bat coronavirus, which we detected in 2016 in bats in a cave in the vicinity of the index pig farm. This new virus (SADS-CoV) originated from the same genus of horseshoe bats (Rhinolophus) as SARS-CoV.
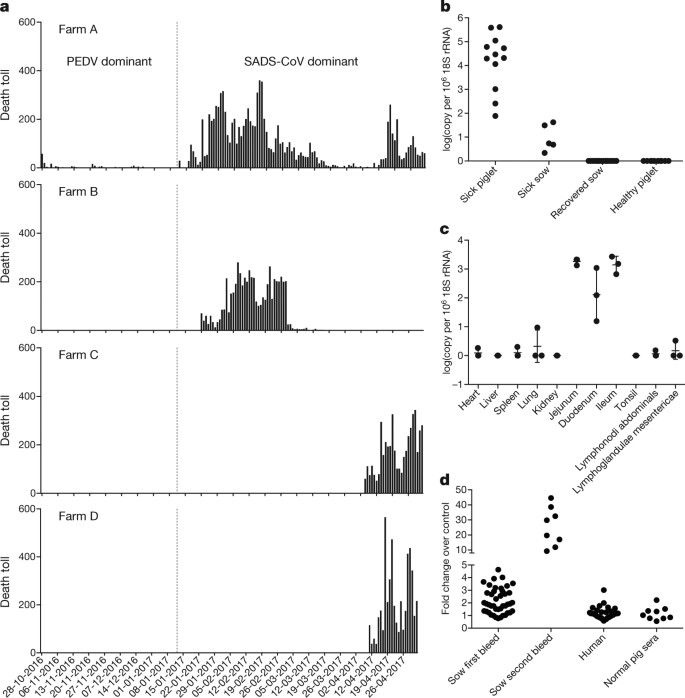
From 28 October 2016 onwards, a fatal swine disease outbreak was observed in a pig farm in Qingyuan, Guangdong province, China, very close to the location of the first known index case of SARS in 2002, who lived in Foshan (Extended Data Fig. 1a). Porcine epidemic diarrhoea virus (PEDV, a coronavirus) had caused prior outbreaks at this farm, and was detected in the intestines of deceased piglets at the start of the outbreak. However, PEDV could no longer be detected in deceased piglets after 12 January 2017, despite accelerating mortality (Fig. 1a), and extensive testing for other common swine viruses yielded no results (Extended Data Table 1). These findings suggested that this was an outbreak of a novel disease. Clinical signs are similar to those caused by other known swine enteric coronaviruses17, 18 and include severe and acute diarrhoea and acute vomiting, leading to death due to rapid weight loss in newborn piglets that are less than five days of age. Infected piglets died 2–6 days after disease onset, whereas infected sows suffered only mild diarrhoea and most sows recovered within two days. The disease caused no signs of febrile illness in piglets or sows. The mortality rate was as high as 90% in piglets that were five days or younger, whereas in piglets that were older than eight days, the mortality dropped to 5%. Subsequently, SADS-related outbreaks were found in three additional pig farms within 20–150 km of the index farm (Extended Data Fig. 1a) and, by 2 May 2017, the disease had caused the death of 24,693 piglets at these four farms (Fig. 1a). In farm A alone, 64% (4,659 out of 7,268) of all piglets that were born in February died. The outbreak has abated, and measures that were taken to control SADS included separation of sick sows and piglets from the rest of the herd. A qPCR test described below was used as the main diagnostic tool to confirm SADS-CoV infection.
Fig. 1: Detection of SADS-CoV infection in pigs in Guangdong, China.
a, Records of daily death toll on the four farms from 28 October 2016 to 2 May 2017. b, Detection of SADS-CoV by qPCR. The y axis shows the log(copy number per 106 copies of 18S rRNA). n = 12 sick piglets, 5 sick sows, 16 recovered sows and 10 healthy piglets. c, Tissue distribution of SADS-CoV in diseased pigs. n = 3. Data are mean ± s.d.; dots represent individual values. d, Detection of SADS-CoV antibodies. n = 46 sows from whom serum was first taken in the first three weeks of the outbreak (First bleed), n = 8 sows from whom serum was taken again (Second bleed) at more than one month after the onset of the outbreak, n = 8 sera from healthy pig controls, n = 35 human sera from pig farmers.
A sample collected from the small intestine of a diseased piglet was analysed by metagenomics analysis using next-generation sequencing (NGS) to identify potential aetiological agents. Of the 15,256,565 total reads obtained, 4,225 matched sequences of the bat CoV HKU2, which was first detected in Chinese horseshoe bats in Hong Kong and Guangdong province, China19. By de novo assembly and targeted PCR, we obtained a 27,173-bp CoV genome that shared 95% sequence identity to HKU2-CoV (GenBank accession number NC_009988). Thirty-three full genome sequences of SADS-CoV were subsequently obtained (8 from farm A, 5 from farm B, 11 from farm C and 9 from farm D) that were 99.9% identical to each other (Supplementary Table 1).
Using qPCR targeting the nucleocapsid gene (Supplementary Table 2), we detected SADS-CoV in acutely sick piglets and sows, but not in recovered or healthy pigs on the four farms, nor in nearby farms that showed no evidence of SADS. The virus replicated to higher titres in piglets than in sows (Fig. 1b). SADS-CoV displayed tissue tropism of the small intestine (Fig. 1c), as observed for other swine enteric coronaviruses20. Retrospective PCR analysis revealed that SADS-CoV was present on farm A during the PEDV epidemic, where the first strongly positive SADS-CoV sample was detected on 6 December 2016. From mid-January onwards, SADS-CoV was the dominant viral agent detected in diseased animals (Extended Data Fig. 1b). It is possible that the presence of PEDV early in the SADS-CoV outbreak may have somehow facilitated or enhanced spillover and amplification of SADS. However the fact that the vast majority of piglet mortality occurred after PEDV infection had become undetectable suggests that SADS-CoV itself causes a lethal infection in pigs that was responsible for these large-scale outbreaks, and that PEDV does not directly contribute to its severity in individual pigs. This was supported by the absence of PEDV and other known swine diarrhoea viruses during the peak and later phases of the SADS outbreaks in the four farms (Extended Data Table 1).
We rapidly developed an antibody assay based on the S1 domain of the spike (S) protein using a luciferase immunoprecipitation system21. Because SADS occurs acutely and has a rapid onset in piglets, serological investigation was conducted only in sows. Among 46 recovered sows tested, 12 were seropositive for SADS-CoV within three weeks of infection (Fig. 1d). To investigate possible zoonotic transmission, serum samples from 35 farm workers who had close contact with sick pigs were also analysed using the same luciferase immunoprecipitation system approach and none were positive for SADS-CoV.
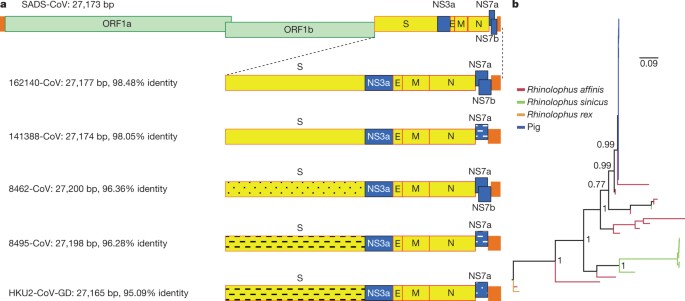
Although the overall genome identity of SADS-CoV and HKU2-CoV is 95%, the S gene sequence identity is only 86%, suggesting that the previously reported HKU2-CoV is not the direct progenitor of SADS-CoV, but that they may have originated from a common ancestor. To test this hypothesis, we developed a SADS-CoV-specific qPCR assay based on its RNA-dependent RNA polymerase (RdRp) gene (Supplementary Table 2) and screened 591 bat anal swabs collected between 2013 and 2016 from seven different locations in Guangdong province (Extended Data Fig. 1a). A total of 58 samples (9.8%) tested positive (Extended Data Table 2), all were from Rhinolophus spp. bats that are also the natural reservoir hosts of SARS-related coronaviruses3,4,5,6,7,8,9,10. Four complete genome sequences with the highest RdRp PCR-fragment sequence identity to that of SADS-CoV were determined by NGS. They are very similar in size (27.2 kb) compared to SADS-CoV (Fig. 2a) and we tentatively call them SADS-related coronaviruses (SADSr-CoV). Overall sequence identity between SADSr-CoV and SADS-CoV ranges from 96 to 98%. Most importantly, the S protein of SADS-CoV shared more than 98% sequence identity with sequences of two of the SADSr-CoVs (samples 162149 and 141388), compared to 86% with HKU2-CoV. The major sequence differences among the four SADSr-CoV genomes were found in the predicted coding regions of the S and NS7a and NS7b genes (Fig. 2a). In addition, the coding region of the S protein N-terminal (S1) domain was determined from 19 bat SADSr-CoVs to enable more detailed phylogenetic analysis.
Fig. 2: Genome and phylogenetic analysis of SADS-CoV and SADSr-CoV.
a, Genome organization and comparison. Colour-coding for different genomic regions as follows. Green, non-structural polyproteins ORF1a and ORF1b; yellow, structural proteins S, E, M and N; blue, accessory proteins NS3a, NS7a and NS7b; Orange, untranslated regions. The level of sequence identity of SADSr-CoV to SADS-CoV is illustrated by different patterns of boxes: Solid colour, highly similar; Dotted fill, moderately similar; Dashed fill, least similar. b, Phylogenetic analysis of 57 S1 sequences (33 from SADS-CoV and 24 from SADSr-CoV). Different colours represent different host species as shown on the left. Scale bar, nucleotide substitutions per site.
The phylogeny of S1 and the full-length genome revealed a high genetic diversity of alphacoronaviruses among bats and strong coevolutionary relationships with their hosts (Fig. 2b and Extended Data Fig. 2), and showed that SADS-CoVs were more closely related to SADSr-CoVs from Rhinolophus affinis than from Rhinolophus sinicus, in which HKU2-CoV was found. Both phylogenetic and haplotype network analyses demonstrated that the viruses from the four farms probably originated from their reservoir hosts independently (Extended Data Fig. 3), and that a few viruses might have undergone further genetic recombination (Extended Data Fig. 4). However, molecular clock analysis of the 33 SADS-CoV genome sequences failed to establish a positive association between sequence divergence and sampling date. Therefore, we speculate that either the virus was introduced into pigs from bats multiple times, or that the virus was introduced into pigs once, but subsequent genetic recombination disturbed the molecular clock.
For viral isolation, we tried to culture the virus in a variety of cell lines (see Methods for details) using intestinal tissue homogenates as starting material. Cytopathogenic effects were observed in Vero cells only after five passages (Extended Data Fig. 5a, b). The identity of SADS-CoV was verified in Vero cells by immunofluorescence microscopy (Extended Data Fig. 5c, d) and by whole-genome sequencing (GenBank accession number MG557844). Similar results were obtained by other groups22, 23.
Known coronavirus host cell receptors include angiotensin-converting enzyme 2 (ACE2) for SARS-related CoV, aminopeptidase N (APN) for certain alphacoronaviruses, such as human (H)CoV-229E, and dipeptidyl peptidase 4 (DPP4) for Middle East respiratory syndrome (MERS)-CoV24,25,26. To investigate the receptor usage of SADS-CoV, we tested live or pseudotyped SADS-CoV infection on HeLa cells that expressed each of the three molecules. Whereas the positive control worked for SARS-related CoV and MERS-CoV pseudoviruses, we found no evidence of enhanced infection or entry for SADS-CoV, suggesting that none of these receptors functions as a receptor for virus entry for SADS-CoV (Extended Data Table 3).
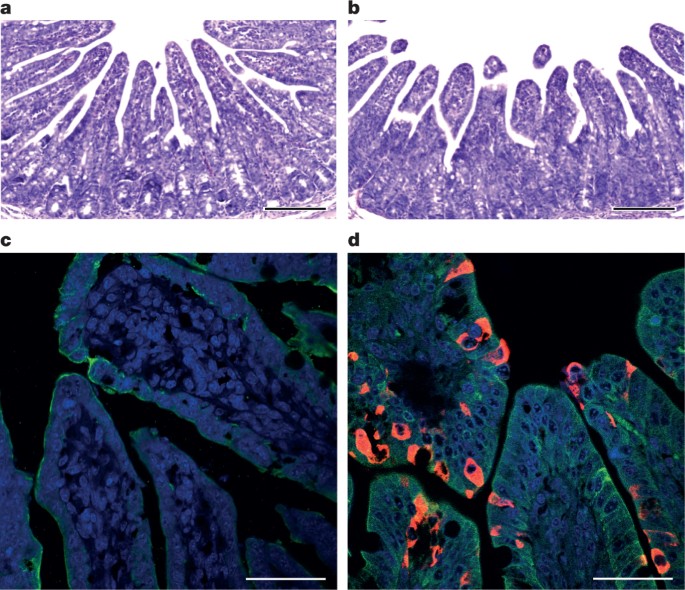
To fulfill Koch’s postulates for SADS-CoV, two different types of animal challenge experiments were conducted (see Methods for details). The first challenge experiment was conducted with specific pathogen-free piglets that were infected with a tissue homogenate of SADS-CoV-positive intestines. Two days after infection, 3 out of 7 animals died in the challenge group whereas 4 out of 5 survived in the control group. Incidentally, the one piglet that died in the control group was the only individual that did not receive colostrum due to a shortage in the supply. It is thus highly likely that lack of nursing and inability to access colostrum was responsible for the death (Extended Data Table 4). For the second challenge, healthy piglets were acquired from a farm in Guangdong that had been free of diarrheal disease for a number of weeks before the experiment, and were infected with the cultured isolate of SADS-CoV or tissue-culture medium as control. Of those inoculated with SADS-CoV, 50% (3 out of 6) died between 2 and 4 days after infection, whereas all control animals survived (Extended Data Table 5). All animals in the infected group suffered watery diarrhoea, rapid weight loss and intestinal lesions (determined after euthanasia upon experiment termination, Extended Data Tables 4, 5). Histopathological examination revealed marked villus atrophy in SADS-CoV inoculated farm piglets four days after inoculation but not in control piglets (Fig. 3a, b) and viral N protein-specific staining was observed mainly in small intestine epithelial cells of the inoculated piglets (Fig. 3c, d).
Fig. 3: Immunohistopathology of SADS-CoV infected tissues.
a–d, Sections of jejunum tissue from control (a, c) and infected (b, d) farm piglets four days after inoculation were stained with haematoxylin and eosin (a, b) or rabbit anti-SADSr-CoV N serum (red), DAPI (blue) and mouse antibodies against epithelial cell markers cytokeratin 8, 18 and 19 (green) in (c, d). SADS-CoV N protein is evident in epithelial cells and deeper in the tissue of infected piglets, which exhibit villus shortening. Scale bars, 200 μm (a, b) and 50 μm (c, d). The experiment was conducted three times independently with similar results.
The current study highlights the value of proactive viral discovery in wildlife, and targeted surveillance in response to an emerging infectious disease event, as well as the disproportionate importance of bats as reservoirs of viruses that threaten veterinary and public health1. It also demonstrates that by using modern technological platforms, such as NGS, luciferase immunoprecipitation system serology and phylogenetic analysis, key experiments that traditionally rely on the isolation of live virus can be performed rapidly before virus isolation.
Methods
Sample collection
Bats were captured and sampled in their natural habitat in Guangdong province (Extended Data Fig. 1) as described previously4. Faecal swab samples were collected in viral transport medium (VTM) composed of Hank’s balanced salt solution at pH 7.4 containing BSA (1%), amphotericin (15 μg ml−1), penicillin G (100 units ml−1) and streptomycin (50 μg ml−1). Stool samples from sick pigs were collected in VTM. When appropriate and feasible, intestinal samples were also taken from deceased animals. Samples were aliquoted and stored at –80 °C until use. Blood samples were collected from recovered sows and workers on the farms who had close contact with sick pigs. Serum was separated by centrifugation at 3,000g for 15 min within 24 h of collection and preserved at 4 °C. Human serum collection was approved by the Medical Ethics Committee of the Wuhan School of Public Health, Wuhan University and Hummingbird IRB. Human, pigs and bats were sampled without gender or age preference unless indicated (for example, piglets or sows). No statistical methods were used to predetermine sample size.
Virus isolation
The following cells were used for virus isolation in this study: Vero (cultured in DMEM and 10% FBS); Rhinolophus sinicus primary or immortalized cells generated in our laboratory (all cultured in DMEM/F12 and 15% FBS): kidney primary cells (RsKi9409), lung primary cells (RsLu4323), lung immortalized cells (RsLuT), brain immortalized cells (RsBrT) and heart immortalized cells (RsHeT); and swine cell lines: two intestinal porcine enterocytes cell lines, IPEC (RPMI1640 and 10% FBS) and SIEC (DMEM and 10% FBS), three kidney cell lines PK15, LLC-PK1 (DMEM and 10% FBS for both) and IBRS (MEM and 10% FBS), and one pig testes cell line, ST (DMEM and 10% FBS). All cell lines were tested free of mycoplasma contamination, species were confirmed and authenticated by microscopic morphologic evaluation. None of the cell lines was on the list of commonly misidentified cell lines (by the ICLAC).
Cultured cell monolayers were maintained in their respective medium. PCR-positive pig faecal samples or the supernatant from homogenized pig intestine (in 200 μl VTM) were spun at 8,000g for 15 min, filtered and diluted 1:2 with DMEM supplemented with 16 μg ml−1 trypsin before addition to the cells. After incubation at 37 °C for 1 h, the inoculum was removed and replaced with fresh culture medium containing antibiotics (below) and 16 μg ml−1 trypsin. The cells were incubated at 37 °C and observed daily for cytopathic effect (CPE). Four blind passages (three-day interval between every passage) were performed for each sample. After each passage, both the culture supernatant and cell pellet were examined for the presence of virus by RT–PCR using the SADS-CoV primers listed in Supplementary Table 2. Penicillin (100 units ml−1) and streptomycin (15 μg ml−1) were included in all tissue culture media.
RNA extraction, S1 gene amplification and qPCR
Whenever commercial kits were used, the manufacturer’s instructions were followed without modification. RNA was extracted from 200 μl of swab samples (bat), faeces or homogenized intestine (pig) with the High Pure Viral RNA Kit (Roche). RNA was eluted in 50 μl of elution buffer and used as the template for RT–PCR. Reverse transcription was performed using the SuperScript III kit (Thermo Fisher Scientific).
To amplify S1 genes from bat samples, nested PCR was performed with primers designed based on HKU2-CoV (GenBank accession number NC_009988.1)19 (Supplementary Table 2). The 25-μl first-round PCR mixture contained 2.5 μl 10× PCR reaction buffer, 5 pmol of each primer, 50 mM MgCl2, 0.5 mM dNTP, 0.1 μl Platinum Taq Enzyme (Thermo Fisher Scientific) and 1 μl cDNA. The 50-μl second-round PCR mixture was identical to the first-round PCR mixture except for the primers. Amplification of both rounds was performed as follows: 94 °C for 5 min followed by 60 cycles at 94 °C for 30 s, 50 °C for 40 s, 72 °C for 2.5 min, and a final extension at 72 °C for 10 min. PCR products were gel-purified and sequenced.
For qPCR analysis, primers based on SADS-CoV RdRp and N genes were used (Supplementary Table 2). RNA extracted from above was reverse-transcribed using PrimeScript RT Master Mix (Takara). The 10 μl qPCR reaction mix contained 5 μl 2× SYBR premix Ex TaqII (Takara), 0.4 μM of each primer and 1 μl cDNA. Amplification was performed as follows: 95 °C for 30 s followed by 40 cycles at 95 °C for 5 s, 60 °C for 30 s, and a melting curve step.
Luciferase immunoprecipitation system assay
The SADS-CoV S1 gene was codon-optimized for eukaryotic expression, synthesized (GenScript) and cloned in frame with the Renilla luciferase gene (Rluc) and a Flag tag in the pREN2 vector21. pREN2-S1 plasmids were transfected into Cos-1 cells using Lipofectamine 2000 (Thermo Fisher Scientific). At 48 h post-transfection, cells were collected, lysed and a luciferase assay was performed to determine Rluc expression for both the empty vector (pREN2) and the pREN2-S1 construct. For testing of unknown pig or human serum samples, 1 μl of serum was incubated with 10 million units of Rluc alone (vector) or Rluc-S1, respectively, together with 3.5 μl of a 30% protein A/G UltraLink resin suspension (Pierce, Thermo Fisher Scientific). After extensive washing to remove unbounded luciferase-tagged antigens, the captured luciferase amount was determined using the commercial luciferase substrate kit (Promega). The ratio of Rluc-S1:Rluc (vector) was used to determine the specific S1 reactivity of pig and human sera. Commercial Flag antibody (Thermo Fisher Scientific) was used as the positive control, and various pig sera (from uninfected animals in China or Singapore; or pigs infected with PEDV, TGEV or Nipah virus) were used as a negative control.
Protein expression and antibody production
The N gene from SADSr-CoV 3755 (GenBank accession number MF094702), which shares a 98% amino acid sequence identity to the SADS-CoV N protein, was inserted into pET-28a+ (Novagen) for prokaryotic expression. Transformed Escherichia coli were grown at 37 °C for 12–18 h in medium containing 1 mM IPTG. Bacteria were collected by centrifugation and resuspended in 30 ml of 5 mM imidazole and lysed by sonication. The lysate, from which N protein expression was confirmed with an anti-His-tag antibody, was applied to Ni2+ resin (Thermo Fisher Scientific). The purified N protein, at a concentration of 400 μg ml−1, was used to immunize rabbits for antibody production following published methods27. After immunization and two boosts, rabbits were euthanized and sera were collected. Rabbit anti-N protein serum was used 1:10,000 for subsequent western blots.
Amplification, cloning and expression of human and swine genes
Construction of expression clones for human ACE2 in pcDNA3.1 has been described previously5, 28. Human DPP4 was amplified from human cell lines. Human APN (also known as ANPEP) was commercially synthesized. Swine APN (also known as ANPEP), DPP4 and ACE2 were amplified from piglet intestine. Full-length gene fragments were amplified using specific primers (provided upon request). Human ACE2 was cloned into pCDNA3.1 fused with a His tag. Human APN and DPP4, swine APN, DPP4 and ACE2 were cloned into pCAGGS fused with an S tag. Purified plasmids were transfected into HeLa cells. After 24 h, expression human or swine genes in HeLa cells was confirmed by immunofluorescence assay using mouse anti-His tag or mouse anti-S tag monoclonal antibodies (produced in house) followed by Cy3-labelled goat anti-mouse/rabbit IgG (Proteintech Group).
Pseudovirus preparation
The codon-humanized S genes of SADS-CoV or MERS-CoV cloned into pcDNA3.1 were used for pseudovirus construction as described previously5, 28. In brief, 15 μg of each pHIV-Luc plasmid (pNL4.3.Luc.R-E-Luc) and the S-protein-expressing plasmid (or empty vector control) were co-transfected into 4 × 106 HEK293T cells using Lipofectamine 3000 (Thermo Fisher Scientific). After 4 h, the medium was replaced with fresh medium. Supernatants were collected 48 h after transfection and clarified by centrifugation at 3,000g, then passed through a 0.45-μm filter (Millipore). The filtered supernatants were stored at −80 °C in aliquots until use. To evaluate the incorporation of S proteins into the core of HIV virions, pseudoviruses in supernatant (20 ml) were concentrated by ultracentrifugation through a 20% sucrose cushion (5 ml) at 80,000g for 90 min using a SW41 rotor (Beckman). Pelleted pseudoviruses were dissolved in 50 μl phosphate-buffered saline (PBS) and examined by electron microscopy.
Pseudovirus infection
HeLa cells transiently expressing APN, ACE2 or DPP4 were prepared using Lipofectamine 2000 (Thermo Fisher Scientific). Pseudoviruses prepared above were added to HeLa cells overexpressing APN, ACE2 or DPP4 24 h after transfection. The unabsorbed viruses were removed and replaced with fresh medium at 3 h after infection. The infection was monitored by measuring the luciferase activity conferred by the reporter gene carried by the pseudovirus, using the Luciferase Assay System (Promega) as follows: cells were lysed 48 h after infection, and 20 μl of the lysates was taken for determining luciferase activity after the addition of 50 μl of luciferase substrate.
Examination of known CoV receptors for SADS-CoV entry/infection
HeLa cells transiently expressing APN, ACE2 or DPP4 were prepared using Lipofectamine 2000 (Thermo Fisher Scientific) in a 96-well plate, with mock-transfected cells as controls. SADS-CoV grown in Vero cells was used to infect HeLa cells transiently expressing APN, ACE2 or DPP4. The inoculum was removed after 1 h of absorption and washed twice with PBS and supplemented with medium. SARS-related-CoV WIV167 and MERS-CoV HIV-pseudovirus were used as positive control for human/swine ACE2 or human/swine DPP4, respectively. After 24 h of infection, cells were washed with PBS and fixed with 4% formaldehyde in PBS (pH 7.4) for 20 min at room temperature. SARS-related-CoV WIV16 replication was detected using rabbit antibody against the SARS-related-CoV Rp3 N protein (made in house, 1:100) followed by Cy3-conjugated goat anti-rabbit IgG (1:50, Proteintech)7. SADS-CoV replication was monitored using rabbit antibody against the SADSr-CoV 3755 N protein (made in house, 1:50) followed by FITC-conjugated goat anti-rabbit IgG (1:50, Proteintech). Nuclei were stained with DAPI (Beyotime). Staining patterns were examined using confocal microscopy on a FV1200 microscope (Olympus). Infection of MERS-CoV HIV-pseudovirus was monitored by luciferase 48 h after infection.
High-throughput sequencing, pathogen screening and genome assembly
Tissue from the small intestine of deceased pigs was homogenized and filtered through 0.45-μm filters before nucleic acid extraction and ribosomal RNA was depleted using the NEBNext rRNA Depletion Kit (New England Biolabs). Metagenomics analysis of both RNA and DNA viruses was performed. For RNA virus screening, the sequencing library was constructed using Ion Total RNA-Seq Kit v2 (Thermo Fisher Scientific). For DNA virus screening, NEBNext Fast DNA Fragmentation & Library Prep Set for Ion Torrent (New England Biolabs) was used for library preparation. Both libraries were sequenced on an Ion S5 sequencer (Thermo Fisher Scientific). An analysis pipeline was applied to the sequencing data, which included the following analysis steps: (1) raw data quality filtering; (2) host genomic sequence filtering; (3) BLASTn search against the virus nucleotide database using BLAST; (4) BLASTx search against the virus protein database using DIAMOND v.0.9.0; (5) contig assembling and BLASTx search against the virus protein database. For whole viral genome sequencing, amplicon primers (provided upon request) were designed using the Thermo Fisher Scientific online tool with the HKU2-CoV and the SADS-CoV farm A genomes as references, and the sequencing libraries were constructed using NEBNext Ultra II DNA Library Prep Kit for Illumina and sequenced on an MiSeq sequencer. PCR and Sanger sequencing was performed to fill gaps in the genome. Genome sequences were assembled using CLC Genomic Workbench v.9.0. 5′-RACE was performed to determine the 5′-end of the genomes using SMARTer RACE 5′/3′ Kit (Takara). Genomes were annotated using Clone Manager Professional Suite 8 (Sci-Ed Software).
Phylogenetic analysis
SADS-CoV genome sequences and other representative coronavirus sequences (obtained from GenBank) were aligned using MAFFT v.7.221. Phylogenetic analyses with full-length genome, S gene and RdRp were performed using MrBayes v.3.2. Markov chain Monte Carlo was run for 20–50 million steps using the GTR+G+I model (general time reversible model of nucleotide substitution with a proportion of invariant sites and γ-distributed rates among sites). The first 10% was removed as burn-in. The association between phylogenies and phenotypes (for example, host species and farms) was assessed by BaTS beta-build2, with the trees obtained in the previous step used as input. For SADS-CoVs, a median-joining network analysis was performed using PopART v.1.7, with ɛ = 0. Phylogenetic analysis of the 33 full-length SADS-CoV genome sequences was performed using RAxML v.8.2.11, with GTRGAMMA as the nucleotide substitution model and 1,000 bootstrap replicates. The maximum likelihood tree was used to test the molecular clock using TempEst v.1.5. Potential genetic recombination events in our datasets were detected using RDP v.4.72.
Animal infection studies
Experiments were carried out strictly in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The use of animals in this study was approved by the South China Agricultural University Committee of Animal Experiments (approval number 201004152).
Two different animal challenge experiments were conducted. Pigs were used without gender preference. In the first experiment, which was conducted before the virus was isolated, we used three-day old specific pathogen-free (SPF) piglets of the same breeding line, cared for at a SPF facility, fed with colostrum (except one). These piglets were bred and reared to be free of PEDV, CSFV, SIV, PCV2 and PPV infections, and were routinely tested for viral infections using PCR. We also conducted NGS to further confirm that these were animals were free of infection of the above viruses before the animal experiment, and to demonstrate that the animals were free of SADS-CoV infection. The intestinal tissue samples from healthy and diseased animals (intestinal samples excised from euthanized piglets, then ground to make slurry for the inoculum and NGS was performed to confirm no other pig pathogens were found in the samples), were used to feed two groups of 5 (control) and 7 (infection) animals, respectively. For the second experiment, isolated SADS-CoV was used to infect healthy piglets from a farm in Guangdong, which had been free of diarrheal disease for a number of weeks. These piglets were from the same breed as those on SADS-affected farms, to eliminate potential host factor differences and to more accurately reproduce the conditions that occurred during the outbreak in the region. Both groups of piglets were cared for at a known pig disease-free facility. Again, qPCR and NGS were used to make sure that there was no other known swine diarrhoea virus present in the virus inoculum or any of the experimental animals. Two groups (6 for each group) of three-day old piglets were inoculated with SADS-CoV culture supernatant or normal cell culture medium as control. NGS and qPCR were used to confirm that there were no other known swine pathogens in the inoculum.
For both experiments, animals were recorded daily for signs of diseases, such as diarrhoea, weight loss and death. Faecal swabs were collected daily from all animals and screened for known swine diarrhoea viruses by qPCR. Weight loss was calculated as the percentage weight loss compared the original weight at day 0 with a threshold of >5%. It is important to point out that piglets when they are three days old tend to suffer from diarrhoea and weight loss when they are taken away from sows and the natural breast-feeding environment even without infection. At experimental endpoints, piglets were humanely euthanized and necropsies performed. Pictures were taken to record gross pathological changes to the intestines. Ileal, jejunal and duodenal tissues were taken from selected animals and stored at –80 °C for further analysis.
Haematoxylin and eosin and immunohistochemistry analysis
Frozen (–80 °C) small intestinal tissues including duodenum, jejunum and ileum taken from the experimentally infected pigs were pre-frozen at –20 °C for 10 min. Tissues were then embedded in optimal cutting temperature (OCT) compound and cut into 8-μm sections using the Cryotome FSE machine (Thermo Fisher Scientific). Mounted microscope slides were fixed with paraformaldehyde and stained with haematoxylin and eosin for histopathological examination.
For immunohistochemistry analysis, a rabbit antibody raised against the SADSr-CoV 3755 N protein was used for specific staining of SADS-CoV antigen. Slides were blocked by incubating with 10% goat serum (Beyotime) at 37 °C for 30 min, followed by overnight incubation at 4 °C with the rabbit anti-3755 N protein serum (1:1,000) and mouse anti-cytokeratin 8+18+19 monoclonal antibody (Abcam), diluted 1:100 in PBST buffer containing 5% goat serum. After washing, slides were then incubated for 50 min at room temperature with Cy3-conjugated goat-anti-rabbit IgG (Proteintech) and FITC-conjugated goat-anti-mouse IgG (Proteintech), diluted 1:100 in PBST buffer containing 5% goat serum. Slides were stained with DAPI (Beyotime) and observed under a fluorescence microscope (Nikon).
Lau, S. K. et al. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl Acad. Sci. USA 102, 14040–14045 (2005).
He, B. et al. Identification of diverse alphacoronaviruses and genomic characterization of a novel severe acute respiratory syndrome-like coronavirus from bats in China. J. Virol. 88, 7070–7082 (2014).
Yang, X. L. et al. Isolation and characterization of a novel bat coronavirus closely related to the direct progenitor of severe acute respiratory syndrome coronavirus. J. Virol. 90, 3253–3256 (2016).
Wu, Z. et al. ORF8-related genetic evidence for Chinese horseshoe bats as the source of human severe acute respiratory syndrome coronavirus. J. Infect. Dis. 213, 579–583 (2016).
Wang, L. et al. Discovery and genetic analysis of novel coronaviruses in least horseshoe bats in southwestern China. Emerg. Microbes Infect. 6, e14 (2017).
Hu, B. et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 13, e1006698 (2017).
Sun, D., Wang, X., Wei, S., Chen, J. & Feng, L. Epidemiology and vaccine of porcine epidemic diarrhea virus in China: a mini-review. J. Vet. Med. Sci. 78, 355–363 (2016).
Lau, S. K. et al. Complete genome sequence of bat coronavirus HKU2 from Chinese horseshoe bats revealed a much smaller spike gene with a different evolutionary lineage from the rest of the genome. Virology 367, 428–439 (2007).
Burbelo, P. D. et al. Serological diagnosis of human herpes simplex virus type 1 and 2 infections by luciferase immunoprecipitation system assay. Clin. Vaccine Immunol. 16, 366–371 (2009).
Ren, W. et al. Difference in receptor usage between severe acute respiratory syndrome (SARS) coronavirus and SARS-like coronavirus of bat origin. J. Virol. 82, 1899–1907 (2008).
We thank S.-B. Xiao for providing pig cell lines, P. Burbelo for providing the luciferase immunoprecipitation system vector and L. Zhu for enabling the rapid synthesis of the S gene; the WIV animal facilities; J. Min for help with the preparation of the immunohistochemistry samples; and G.-J. Zhu and A. A. Chmura for assistance with bat sampling. This work was jointly supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDPB0301) to Z.-L.S., China Natural Science Foundation (81290341 and 31621061 to Z.-L.S., 81661148058 to P.Z., 31672564 and 31472217 to J.-Y.M., 81572045, 81672001 and 81621005 to Y.-G.T.), National Key Research and Development Program of China (2015AA020108, 2016YFC1202705, AWS16J020 and AWS15J006) to Y.-G.T.; National Science and Technology Spark Program (2012GA780026) and Guangdong Province Agricultural Industry Technology System Project (2016LM1112) to J.-Y.M., State Key Laboratory of Pathogen and Biosecurity (SKLPBS1518) to Y.-G.T., Taishan Scholars program of Shandong province (ts201511056 to W.-F.S.), NRF grants NRF2012NRF-CRP001–056, NRF2016NRF-NSFC002-013 and NMRC grant CDPHRG/0006/2014 to L.-F.W., Funds for Environment Construction & Capacity Building of GDAS’ Research Platform (2016GDASPT-0215) to LBZ, United States Agency for International Development Emerging Pandemic Threats PREDICT project (AID-OAA-A-14-00102), National Institute of Allergy and Infectious Diseases of the National Institutes of Health (Award Number R01AI110964) to P.D. and Z.-L.S.
Reviewer information
Nature thanks C. Drosten, G. Palacios and L. Saif for their contribution to the peer review of this work.
Author information
Author notes
These authors contributed equally: Peng Zhou, Hang Fan, Tian Lan.
Affiliations
CAS Key Laboratory of Special Pathogens and Biosafety, Wuhan Institute of Virology, Chinese Academy of Sciences, Wuhan, China
Peng Zhou
, Xing-Lou Yang
, Wei Zhang
, Yan Zhu
, Xiao-Shuang Zheng
, Bei Li
, Hua Guo
, Yun Luo
, Xiang-Ling Liu
, Jing Chen
& Zheng-Li Shi
Beijing Institute of Microbiology and Epidemiology, Beijing, China
Hang Fan
, Ya-Wei Zhang
, Jin-Man Li
, Guang-Qian Pei
, Xiao-Ping An
, Zhi-Qiang Mi
, Tong-Tong He
, Yong Huang
, Qiang Sun
, Xiang-Li-Lan Zhang
, Yuan-Yuan Wang
, Shao-Zhen Xing
& Yi-Gang Tong
College of Animal Science, South China Agricultural University, Guangzhou, China
Tian Lan
, Qing-Mei Xie
, Jun-Wei Chen
, Ling Zhou
, Kai-Jie Mai
, Zi-Xian Wu
, Di Li
, Yan-Shan Chen
, Yuan Sun
& Jing-Yun Ma
Key Laboratory of Animal Health Aquaculture and Environmental Control, Guangzhou, China
Tian Lan
, Qing-Mei Xie
, Jun-Wei Chen
, Ling Zhou
, Kai-Jie Mai
, Zi-Xian Wu
, Di Li
, Yan-Shan Chen
, Yuan Sun
& Jing-Yun Ma
Key Laboratory of Etiology and Epidemiology of Emerging Infectious Diseases in Universities of Shandong, Taishan Medical College, Taian, China
Wei-Feng Shi
& Juan Li
Programme in Emerging Infectious Diseases, Duke-NUS Medical School, Singapore, Singapore
Shailendra Mani
, Danielle E. Anderson
& Lin-Fa Wang
Guangdong Key Laboratory of Animal Conservation and Resource Utilization, Guangdong Public Laboratory of Wild Animal Conservation and Utilization, Guangdong Institute of Applied Biological Resources, Guangzhou, China
Li-Biao Zhang
School of Public Health, Wuhan University, Wuhan, China
Shi-Yue Li
Guangdong Key Laboratory of Laboratory Animals, Guangdong Laboratory Animals Monitoring Institute, Guangzhou, China
Feng Cong
, Peng-Ju Guo
& Ren Huang
EcoHealth Alliance, New York, NY, USA
Peter Daszak
School of Life Sciences, North China University of Science and Technology, Tangshan, China
Yi-Gang Tong
Contributions
L.-F.W., Z.-L.S., P.Z., Y.-G.T., and J.-Y.M. conceived the study. P.Z., W.Z., Y.Z., S.M., X.-S.Z., B.L., X.-L.Y., H.G., D.E.A., Y.L., X.L.L. and J.C. performed qPCR, serology and histology experiments and cultured the virus. H.F., Y.-W.Z., J.-M.L., G.-Q.P., X.-P.A., Z.-Q.M., T.-T.H., Y.H., Q.S., Y.-Y.W., S.-Z.X., X.-L.-L.Z., W.-F.S. and J.L. performed genome sequencing and annotations. T.L., Q.-M.X., J.-W.C., L.Z., K.-J.M., Z.-X.W., Y.-S.C., D.L., Y.S., F.C., P.-J.G. and R.H. prepared the samples and carried out animal challenge experiments. Z.-L.S., P.D., L.-B.Z., S.-Y.L. coordinated collection of bat samples. P.Z., L.-F.W., Z.-L.S. and P.D. had a major role in the preparation of the manuscript.
a, SADS-affected farms are labelled (farms A–D) with blue swine silhouettes following the temporal sequence of the outbreaks. Bat sampling sites are indicated with black bat silhouettes. The bat SADSr-CoV that is most closely related to SADS-CoV (sample 162140) originated in Conghua. The red flag marks Foshan city, the site of the SARS index case. b, Pooled intestinal samples (n = 5 or more biological independent samples) were collected at dates given on the x axis from deceased piglets and analysed by qPCR. The viral load for each piglet is shown as copy number per milligram of intestine tissue (y axis).
a, Bayesian phylogenetic tree of the full-length genome. b, Bayesian phylogenetic tree of the ORF1a and ORF1b sequences. Trees were constructed using MrBayes with the average standard deviation of split frequencies under 0.01. The host of each sequence is represented as a silhouette. Newly sequenced SADS-CoVs are highlighted in red, bat SADSr-CoVs are shown in blue and previously published sequences are shown in black. Scale bars, nucleotide substitutions per site.
a, Phylogenetic tree constructed using MrBayes. The GTR+GAMMA model was applied and 20 million steps were run, with the first 10% removed as burn in. Viruses from different farms are labelled with different colours. Scale bar, nucleotide substitutions per site. b, Median-joining haplotype network constructed using ProART. In this analysis, ɛ = 0 was used. The size of the circles represents the number of samples. The larger the circle, the more samples it includes.
The potential genetic recombination events were detected using RDP. For each virus strain, different colours represent different sources of the genomes. Source data
a, b, Vero cells are shown 20 h after infection with mock (a) or SADS-CoV (b). c, d, Mock or SADS-CoV-infected samples stained with rabbit serum raised against the recombinant SADSr-CoV N protein (red) and DAPI (blue). The experiment was conducted independently three times with similar results. Scale bars, 100 μm. Source data
Extended Data Table 1 List of all known swine viruses tested by PCR at the beginning of the of SADS outbreak investigation on the four farms
This file contains two supplementary tables. Supplementary table 1 contains a list of nucleotide variants among the 33 SADS-CoV genomes obtained from the four farms. Supplementary table 2 contains a list of PCR primers used in this study
Zhou, P., Fan, H., Lan, T. et al. Fatal swine acute diarrhoea syndrome caused by an HKU2-related coronavirus of bat origin. Nature 556, 255–258 (2018). https://doi.org/10.1038/s41586-018-0010-9
The discovery of SARS-like coronavirus in bats suggests that bats could be the natural reservoir of SARS-CoV. However, previous studies indicated the angiotensin-converting enzyme 2 (ACE2) protein, a known SARS-CoV receptor, from a horseshoe bat was unable to act as a functional receptor for SARS-CoV. Here, we extended our previous study to ACE2 molecules from seven additional bat species and tested their interactions with human SARS-CoV spike protein using both HIV-based pseudotype and live SARS-CoV infection assays. The results show that ACE2s of Myotis daubentoni and Rhinolophus sinicus support viral entry mediated by the SARS-CoV S protein, albeit with different efficiency in comparison to that of the human ACE2. Further, the alteration of several key residues either decreased or enhanced bat ACE2 receptor efficiency, as predicted from a structural modeling study of the different bat ACE2 molecules. These data suggest that M. daubentoni and R. sinicus are likely to be susceptible to SARS-CoV and may be candidates as the natural host of the SARS-CoV progenitor viruses. Furthermore, our current study also demonstrates that the genetic diversity of ACE2 among bats is greater than that observed among known SARS-CoV susceptible mammals, highlighting the possibility that there are many more uncharacterized bat species that can act as a reservoir of SARS-CoV or its progenitor viruses. This calls for continuation and expansion of field surveillance studies among different bat populations to eventually identify the true natural reservoir of SARS-CoV.
Introduction
Severe acute respiratory syndrome coronavirus (SARS-CoV) is the aetiological agent responsible for the SARS outbreaks during 2002–2003, which had a huge global impact on public health, travel and the world economy [4, 11]. The host range of SARS-CoV is largely determined by the specific and high-affinity interactions between a defined receptor-binding domain (RBD) on the SARS-CoV spike protein and its host receptor, angiontensin-converting enzyme 2 (ACE2) [6, 7, 9]. It has been hypothesized that SARS-CoV was harbored in its natural reservoir, bats, and was transmitted directly or indirectly from bats to palm civets and then to humans [10]. However, although the genetically related SARS-like coronavirus (SL-CoV) has been identified in horseshoe bats of the genus Rhinolophus [5, 8, 12, 18], its spike protein was not able to use the human ACE2 (hACE2) protein as a receptor [13]. Close examination of the crystal structure of human SARS-CoV RBD complexed with hACE2 suggests that truncations in the receptor-binding motif (RBM) region of SL-CoV spike protein abolish its hACE2-binding ability [7, 10], and hence the SL-CoV found recently in horseshoe bats is unlikely to be the direct ancestor of human SARS-CoV. Also, it has been shown that the human SARS-CoV spike protein and its closely related civet SARS-CoV spike protein were not able to use a horseshoe bat (R. pearsoni) ACE2 as a receptor [13], highlighting a critical missing link in the bat-to-civet/human transmission chain of SARS-CoV.
There are at least three plausible scenarios to explain the origin of SARS-CoV. First, some unknown intermediate hosts were responsible for the adaptation and transmission of SARS-CoV from bats to civets or humans. This is the most popular theory of SARS-CoV transmission at the present time [10]. Second, there is an SL-CoV with a very close relationship to the outbreak SARS-CoV strains in a non-bat animal host that is capable of direct transmission from reservoir host to human or civet. Third, ACE2 from yet to be identified bat species may function as an efficient receptor, and these bats could be the direct reservoir of human or civet SARS-CoV. Unraveling which scenario is most likely to have occurred during the 2002–2003 SARS epidemic is critical for our understanding of the dynamics of the outbreak and will play a key role in helping us to prevent future outbreaks. To this end, we have extended our studies to include ACE2 molecules from different bat species and examined their interaction with the human SARS-CoV spike protein. Our results show that there is great genetic diversity among bat ACE2 molecules, especially at the key residues known to be important for interacting with the viral spike protein, and that ACE2s of Myotis daubentoni and Rhinolophus sinicus from Hubei province can support viral entry.
Materials and methods
Cell lines and antibodies
HeLa cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (Gibco, USA). Rabbit polyclonal antibodies against ACE2 of R. pearsoni (RpACE2) were generated using R. pearsoni ACE2 protein expressed in Escherichia coli at the Wuhan Institute of Virology following standard procedures.
Bat sample collection and identification
Bats were sampled from their natural habitats in Hubei, Guangxi, Guizhou, Henan and Yunnan provinces in China as described previously [8]. Bat identification was initially determined in the field by morphology and later confirmed in the laboratory by sequencing the mitochondrial cytochrome b gene from samples of blood cells or rectal tissue as described previously [1].
Bat ACE2 amplification and cloning
Total RNA was extracted from bat rectal tissue using TRIzol Reagent (Invitrogen, USA) and treating with RNase-free DNase I at 37°C for 30 min. First-strand cDNA was synthesized from total RNA by reverse transcription with random hexamers. Full-length bat ACE2 fragments were amplified using the forward primer bAF2 (5′-CTTGGTACCATGTCAGGCTCTTYCTGG-3′) and the reverse primer bAR2 (5′-CCGCTCGAGCTAAAAB[G/T/C]GAV[G/A/C]GTCTGAACATCATC-3′). The PCR mixture (25 μL) contained 0.5 μL cDNA, 1.5 mM MgCl2 and 0.2 μM of each primer, and the fragments were amplified using the following parameters: 95°C for 5 min, 35 cycles of 94°C for 30 s, 55°C for 45 s and 68°C for 3 min, with a final elongation step at 68°C for 10 min. All bat ACE2s were cloned into pCDNA3.1 with KpnI and XhoI, and this was verified by sequencing.
Chimeric ACE2 construction
For samples in which full-length ACE2 amplification was unsuccessful, the N-terminal region (1–1,170 bp) was amplified using the forward primer bAF2 and the reverse primer RMR (5′-TTAGCTCCATTTCTTAGCAGGTAGG-3′). Chimeric ACE2 was constructed by combining the N-terminal region of bat ACE2 with the C-terminal portion of human ACE2 at the unique BamHI site (1,070–1,075 bp). The chimera was subsequently cloned into pCDNA3.1 with KpnI and XhoI and sequenced as above.
Construction of bat ACE2 mutants
ACE2 from M. daubentoni was chosen to generate a series of ACE2 mutants using a QuikChange II Site-Directed Mutagenesis Kit (Stratagene, USA). The altered amino acid codon for each mutant is indicated as follows: I27T, N31K, K35E, and H41Y. Mutants were confirmed by sequencing.
Sequence analysis
All bat ACE2s were submitted to GenBank (EF569964, GQ999931–GQ999938). Sequence alignment was performed using ClustalX version 1.83 [15] and corrected manually. A phylogenetic tree based on amino acid (aa) sequences was constructed using the neighbor-joining (NJ) method in MEGA version 4.1. [14].
Analysis of ACE2 expression by western blotting
Lysates of HeLa cells expressing human ACE2 or bat ACE2 were separated on a 4–10% SDS-PAGE gel, followed by transfer to a polyvinylidene difluoride (PVDF) membrane using a semi-dry protein transfer apparatus (Bio-Rad, USA). The membrane was probed with a rabbit polyclonal antibody against the bat ACE2 protein (1:200) at room temperature for 1 h, followed by incubation with alkaline-phosphatase-conjugated goat anti-rabbit IgG (1:1,000) (Chemicon, Australia). The probed proteins were visualized using NBT and BCIP color development (Promega, USA).
Pseudotype virus infection assays
An HIV-1-luciferase pseudotype virus carrying the SARS-CoV BJ01 S protein, HIV/BJ01-S, was prepared as described previously [13]. HeLa cells were seeded onto 96-well plates for 18 h and then transfected with 0.2 μg recombinant plasmid containing bat or human ACE2 using 0.5 μL Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer’s protocol. At 24 h post-transfection, 30 μL medium containing HIV/BJ01-S was added to each well. At 2–3 h postinfection, unadsorbed viruses were removed, and fresh medium was added. The infection was monitored by measuring luciferase activity, expressed from the reporter gene carried by the pseudovirus, using a luciferase assay kit (Promega, USA). Cells were lysed at 48 h postinfection by adding 20 μL lysis buffer provided with the kit, and 10 μL of the resulting lysates were tested for luciferase activity by the addition of 20 μL luciferase substrate in a Turner Designs TD-20/20 luminometer. Each infection experiment was conducted in triplicate, and all experiments were repeated three times.
Live virus infection assays
Live SARS-CoV infection was carried out under BSL4 conditions at the Australian Animal Health Laboratory (AAHL) as described previously [16, 17]. Briefly, 48 h after transfection, the time at which expression of the ACE2 receptor on the HeLa cell surface is optimal, 2 × 106 TCID50 of virus was added to the cells for infection. The cells were fixed 24 h later by treatment with 100% methanol for 10 min and washed five times with PBST. The primary antibody, chicken anti-SARS-CoV S (produced against the recombinant S protein expressed in E. coli at AAHL), at a 1:500 dilution in 1% BSA/PBS, was added and incubated with the cells for 1 h at room temperature. An FITC anti-chicken conjugate (Chemicon, Australia) at 1:1,000 in 1% BSA/PBS was added after washing the cells five times and incubated with the cells for 1 h. Infection was monitored by immunofluorescent microscopic analysis.
Results and discussion
Cloning and expression of ACE2 genes from different bat species
ACE2 genes from seven bat species were amplified and cloned (Fig. 1, sFig. 1). Full-length genes were obtained from Rhinolophus ferrumequinum from Hubei province (Rf-HB), R. macrotis from Hubei province (Rm-HB), R. pearsoni from Guangxi (Rp-GX), R. pusillus from Hubei province (Rpu-HB), R. sinicus from Guangxi province (Rs-GX) and R. sinicus from Hubei province (Rs-HB). For the following bat species, amplification of the full-length coding region was not successful, and instead,the N-terminal region was cloned in frame with the C-terminal region of the human ACE2 gene to form a chimeric full-length ACE molecule: R. pearsoni from Guizhou province (Rp-GZ), Myotisdaubentonii bat from Yunnan province (Md-YN) and Hipposideros pratti bat from Henan province (Hp-HN). The full-length sequences of bat ACE2 are identical in size to that of hACE2 (805 aa in total). Sequence comparison showed that bat ACE2s are closely related to ACE2s of other mammals and have an aa sequence identity of 80–82% to human and civet ACE2. The aa identity of ACE2 from different bat families ranges from 78 to 84%, and within the genus Rhinolophus, the sequence identity increases to 89–98%. The major sequence variation among bat ACE2s is located in the N-terminal region, which has been identified in structural studies as the SARS-CoV-binding region [6, 7]. A phylogenetic tree was constructed based on the sequences of bat ACE2 (sFig. 2) using the MEGA package [14].
Fig. 1
Sequence alignment of SARS-CoV binding regions of ACE2s from 9 bats, civet and human. The GenBank accession numbers of bat, civet and human ACE2 are as follows: human (NM021804), civet (AY881174), Rf-HB (GQ999931), Rm-HB (GQ999932), Rs-GX (GQ999933), Rp-GX (EF569964), Hp-HN (GQ999934), Rp-GZ (GQ999935), Rs-HB (GQ999936), Md-YN(GQ999937) and Rpu-HB (GQ999938). The alignment was generated using ClustalX v1.83. In black are single, fully conserved residues. In gray are strongly conserved residues. In light gray are weakly conserved residues. Asterisks indicate residues that interact directly with the receptor-binding domain of the SARS-CoV S protein
All ACE2 genes were cloned into a eukaryotic expression vector and used to transfect HeLa cells. Western blot analysis showed that all ACE2s were expressed efficiently and at very similar levels and were recognized by a rabbit anti-bat ACE2 antibody with an apparent molecular weight of approximately 100–130 kDa (Fig. 2c).
Fig. 2
Testing of the ability of bat ACE2 proteins to mediate pseudovirus HIV/BJ01-S and live SARS- CoV infection. a HeLa cells transfected with plasmids encoding bat and human ACE2s were infected with pseudovirus HIV/BJ01-S. Infectivity was determined by measuring the activity of reporter luciferase as described in “Materials and methods”. HeLa cells transfected with pcDNA3.1 and human ACE2 were used as the negative and positive controls, respectively. All tests were performed in triplicate, and the experiments were repeated three times. The error bar represents the calculated standard deviation. I27T, N31K, K35E, and H41Y are mutants of MdACE2 that were made using a QuikChange II Site-Directed Mutagenesis Kit. b SARS-CoV live virus infection using the ACE2s from bats as described in “Materials and methods”. HeLa cells transfected with pcDNA3.1 and human ACE2 were used as the negative and positive controls, respectively. c Expression of bat or human ACE2. Lysates from HeLa cells transfected with plasmid expressing human or bat ACE2 were analyzed by western blot. Rabbit anti-bat ACE2 polyclonal antibody (upper panel) or β-actin monoclonal antibody (lower panel) was used as the primary antibody. Lane 1 vector pcDNA3.1 control; lanes 2–10 bat ACE2 from samples Rf-HB, Rm-HB, Rpu-HB, Hp-HN, Rp-HB, Rp-GZ, Rs-GX, Rs-HB and Md-YN; lanes 11–14 Md-YN ACE2 mutant I27T, N31K, K35E and H41Y; lane 15 human ACE2. The abbreviations of bat species are given in the main text
Functionality of bat ACE2 as an SARS-CoV entry receptor
To examine the susceptibility of different bat ACE2 molecules to SARs-CoV entry, the HIV/BJ01-S pseudovirus system was used to infect HeLa cells transiently expressing bat ACE2 or human ACE2 genes. Among the bat ACE2s, only MdACE2 (MdACE2) and Rs-HB ACE2 demonstrated significant pseudovirus infection, as deduced from the significantly higher level of luciferase activity in comparison to background activity in the negative control (Fig. 2a). Although such assays are not to be viewed as an absolute quantification of receptor activity, it is nevertheless worth noting that MdACE2-mediated infection seemed to be more efficient than with Rs-HB ACE2. In the same context, it is clear that the bat ACE2s were less efficient overall than the human ACE2 in this particular assay system. The biological significance of this observation remains to be determined. The functionality of MdACE2 and Rs-HB ACE2 as SARS-CoV entry receptors was further confirmed by infection with live virus. As shown in Fig. 2b, both bat ACE2 proteins could clearly support SARs-CoV infection. No attempt was made to quantify infection efficiency in this study due to difficulties encountered in conducting experiments under BSL4 conditions.
Structural modeling of bat ACE2 molecules
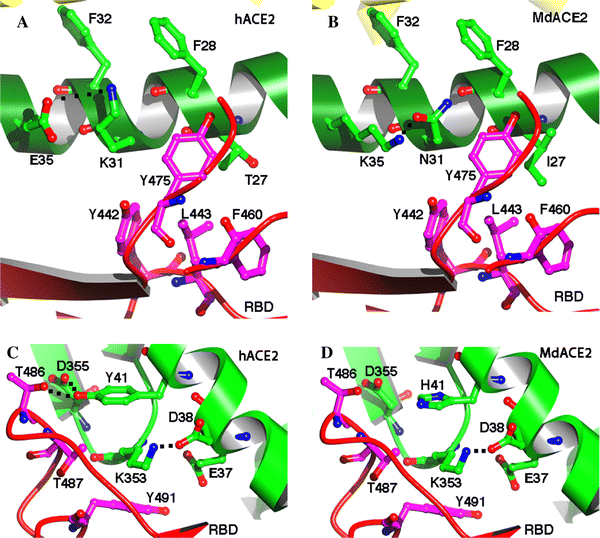
Homologous structural modeling of human SARS-CoV RBD complexed with MdACE2 supports MdACE2 as a receptor for human SARS-CoV S protein. The crystal structure of human SARS-CoV RBD complexed with hACE2 shows that two salt bridges at the SARS-CoV-hACE2 interface, between hACE2 Lys31 and Glu35 and between hACE2 Lys353 and hACE2 Glu38, are both buried in a hydrophobic environment and contribute critically to the SARS-CoV-hACE2 interactions (Fig. 3a, c) [7]. Disturbance of the formation of either of these salt bridges weakens SARS-CoV-hACE2 binding. The Lys31-Glu35 salt bridge at the SARS-CoV-hACE2 interface becomes an Asn31-Lys35 hydrogen bond at the SARS-CoV-Md-YNACE2 interface (Fig. 3b), which possibly weakens virus-receptor binding but still is largely compatible with the virus-receptor interface. Thr27 on hACE2 supports the Lys31-Gu35 salt bridge through hydrophobic interactions with Tyr475 (Fig. 3a); Ile27 on MdACE2 supports the Asn31-Lys35 hydrogen bond more efficiently than Thr27 through tighter hydrophobic interactions with Tyr475 (Fig. 3b). Moreover, Tyr41 on hACE2 supports the Lys353-Glu38 salt bridge (Fig. 3c); His41 on MdACE2 supports the same salt bridge less efficiently than Tyr41 (Fig. 3d). Overall, MdACE2 is an efficient receptor for SARS-CoV, despite the fact that its receptor activity is lower than that of hACE2.
Fig. 3
Homologous structural modeling of SARS-CoV and Md-YN ACE2 (MdACE2) interactions. a Critical salt bridge between hACE2 Lys31 and Glu35 and the hydrophobic residues surrounding it, based on the experimentally determined crystal structure of SARS-CoV RBD complexed with hACE2 (PDB 2AJF). b Homologous structural modeling of the hydrogen bond between MdACE2 Asn31 and Lys35. The modeling was done in the program O [3]. c Critical salt bridge between hACE2 Lys353 and Glu38 and the hydrophobic residues surrounding it, based on the structure of SARS-CoV RBD complexed with hACE2. d Homologous structural modeling of the salt bridge between MdaACE2 Lys353 and SARS-CoV Glu38 and the hydrophobic residues surrounding it. Structural illustrations were prepared using the program Povscript [2]
Compared with MdACE2, Rs-HB ACE2 contains Glu31 and Glu35, which are not compatible with each other due to their same negative charges, which disfavor virus-receptor binding. However, Rs-HB ACE2 also contains Thr27 and Tyr41, both of which support SARS-CoV entry by contributing favorably to the hydrophobic interactions at the virus-receptor interface. Thus, Rs-HB is a low-efficiency receptor for SARS-CoV. All of the other bat ACE2 molecules contain combinations of the aforementioned key residues that are completely incompatible with virus–receptor interactions. More specifically, they either contain same-charged residues at the 31 and 35 positions, which repel each other, or contain His41 and Lys27, which disfavor SARS-CoV binding (Fig. 1). In particular, Lys27 on some of these bat ACE2 molecules is incompatible with certain hydrophobic residues, such as Leu443 and Phe460, on SARS-CoV RBD (Fig. 3a, b). Therefore, these bat ACE2 molecules are not receptors for SARS-CoV.
Site-directed mutagenesis analysis
To confirm the above homologous structural analysis, we carried out site-directed mutagenesis on MdACE2. Our results show that mutations E31K, K35E, and I27T all dramatically decrease the receptor activity of MdACE2, whereas mutation H41Y greatly increases its receptor activity (Fig. 2a). Therefore, our mutagenesis data further confirmed that key residues in ACE2 determine the receptor activity of MdACE2.
Our finding that M. daubentoni and R. sinicus could support SARS-CoV infection has important implications in relation to the origin of SARS-CoV. Since all lines of investigation have indicated that ACE2-binding affinity is among the important determinants for SARS-CoV host range, our data would suggest that M. Daubentonii and R. sinicus have the potential to serve as the direct reservoirs for human SARS-CoV or its highly related civet SARS-CoV. To further investigate the potential of M. Daubentonii and R. sinicus as reservoirs for SARS-CoV, more efforts will have to be directed toward widening the surveillance of bats in these families and in different geographical locations.
Another important finding of our current study is the great genetic diversity of bat ACE2 proteins, which is in contrast to the genetically homogenous hACE2 [10]. Sequence variations of bat ACE2, especially in positions that are critical to SARS-CoV binding, such as residues 27, 31, 35, and 41, suggest that, in addition to the Md-YN and Rs-HB ACE2s, there may be many other bats with an ACE2 protein that makes them susceptible to SARS-CoV entry. This again highlights the need for more field surveillance and molecular characterization of different bat ACE2 proteins until the true reservoir host of SARS-CoV is identified and its spillover mechanisms and transmission pathways are fully characterized.
References
1.
Cui J, Han N, Streicker D, Li G, Tang X, Shi Z, Hu Z, Zhao G, Fontanet A, Guan Y, Wang L, Jones G, Field HE, Daszak P, Zhang S (2007) Evolutionary relationships between bat coronaviruses and their hosts. Emerg Infect Dis 13:1526–1532
Fenn TD, Ringe D, Petsko GA (2003) POVScript+: a program for model and data visualization using persistence of vision ray-tracing. J Appl Crystallogr 36:944–947
Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47:110–119
Lau SK, Woo PC, Li KS, Huang Y, Tsoi HW, Wong BH, Wong SS, Leung SY, Chan KH, Yuen KY (2005) Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc Natl Acad Sci USA 102:14040–14045
Li F (2008) Structural analysis of major species barriers between humans and palm civets for severe acute respiratory syndrome coronavirus infections. J Virol 82:6984–6991
Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, Wang H, Crameri G, Hu Z, Zhang H, Zhang J, McEachern J, Field H, Daszak P, Eaton BT, Zhang S, Wang LF (2005) Bats are natural reservoirs of SARS-like coronaviruses. Science 310:676–679
Li W, Zhang C, Sui J, Kuhn JH, Moore MJ, Luo S, Wong SK, Huang IC, Xu K, Vasilieva N, Murakami A, He Y, Marasco WA, Guan Y, Choe H, Farzan M (2005) Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J 24:1634–1643
Li W, Wong SK, Li F, Kuhn JH, Huang IC, Choe H, Farzan M (2006) Animal origins of the severe acute respiratory syndrome coronavirus: insight from ACE2-S-protein interactions. J Virol 80:4211–4219
Ren W, Li W, Yu M, Hao P, Zhang Y, Zhou P, Zhang S, Zhao G, Zhong Y, Wang S, Wang LF, Shi Z (2006) Full-length genome sequences of two SARS-like coronaviruses in horseshoe bats and genetic variation analysis. J Gen Virol 87:3355–3359
Ren W, Qu X, Li W, Han Z, Yu M, Zhou P, Zhang SY, Wang LF, Deng H, Shi Z (2008) Difference in receptor usage between severe acute respiratory syndrome (SARS) coronavirus and SARS-like coronavirus of bat origin. J Virol 82:1899–1907
Tu C, Crameri G, Kong X, Chen J, Sun Y, Yu M, Xiang H, Xia X, Liu S, Ren T, Yu Y, Eaton BT, Xuan H, Wang LF (2004) Antibodies to SARS coronavirus in civets. Emerg Infect Dis 10:2244–2248
Yu M, Stevens V, Berry JD, Crameri G, McEachern J, Tu C, Shi Z, Liang G, Weingartl H, Cardosa J, Eaton BT, Wang LF (2008) Determination and application of immunodominant regions of SARS coronavirus spike and nucleocapsid proteins recognized by sera from different animal species. J Immunol Methods 331:1–12
Yuan J, Hon CC, Li Y, Wang D, Xu G, Zhang H, Zhou P, Poon LL, Lam TT, Leung FC, Shi Z (2010) Intraspecies diversity of SARS-like coronaviruses in Rhinolophus sinicus and its implications for the origin of SARS coronaviruses in humans. J Gen Virol 91:1058–1062
This work was jointly funded by the State Key Program for Basic Research Grants (2005CB523004, 2010CB530100) from the Chinese Ministry of Science, Technology and the Knowledge Innovation Program Key Project administered by the Chinese Academy of Sciences (KSCX1-YW-R-07) to Z.S. and the CSIRO CEO Science Leader Award to L.-F.W. We thank Gary Crameri and Jennifer Barr for help with live virus infection studies.
Author information
Affiliations
State Key Laboratory of Virology, Wuhan Institute of Virology, Chinese Academy of Sciences (CAS), Wuhan, 430071, Hubei, China
Yuxuan Hou
, Cheng Peng
, Yan Li
, Zhenggang Han
& Zhengli Shi
Australian Animal Health Laboratory, Commonwealth Scientific and Industrial Research Organization Livestock Industries, PO Bag 24, Geelong, VIC, 3220, Australia
Meng Yu
& Lin-Fa Wang
Department of Pharmacology, University of Minnesota Medical School, Minneapolis, MN, 55455, USA
Hou, Y., Peng, C., Yu, M. et al. Angiotensin-converting enzyme 2 (ACE2) proteins of different bat species confer variable susceptibility to SARS-CoV entry. Arch Virol155, 1563–1569 (2010). https://doi.org/10.1007/s00705-010-0729-6